
Methods for Protein Structure Prediction and Function, and Gaseous Protein Folding
An overview of the ways in which we can predict a protein's 3D structure and their function, as well as excerpts from my conversation with Lars Konermann on gaseous protein folding.

Proteins, beyond bodybuilding, are responsible, and often play critical roles, for a majority of biological processes. Each protein molecules consists of one or more chains, made by linking amino acids through a dehydration reaction, where a water molecules is removed for each amino acid bond formed. These chains are then folded, according to the sequence of amino acids, into a distinctive 3D shape. This 3D shape directly influences the actions the protein can perform: where it can bind to other molecules, how it can transport molecules, and how it can be a receptor just to name a few. For example, some proteins form pockets named active sites that perfectly fit to bind a particular target molecule. There is a strong relationship between protein structure and function. You can think of it in the same way as a tool: The shape of a hammer is vital to its function, and altering its shape won’t allow it to hammer nails properly.
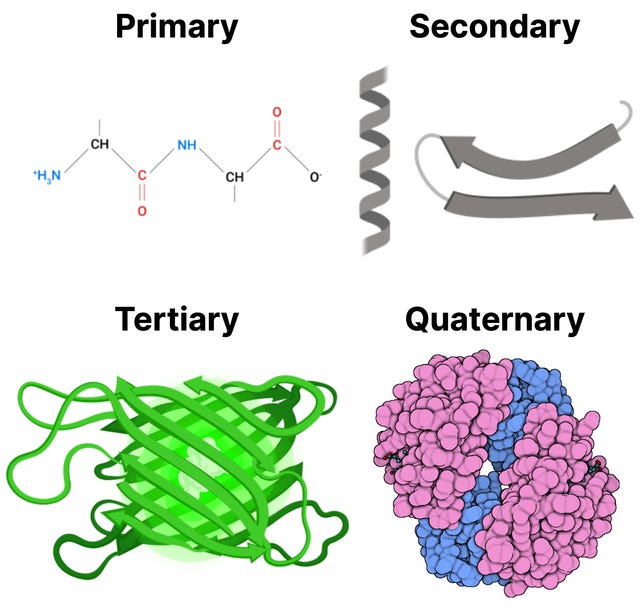
Understanding protein structure is integral to giving us clues to a proteins function. Yet, the question of what a protein does inside a living cell is not as simple as determining the structure. Imagine an analysis of an uncharacterized protein’s structure and amino acid sequence suggests it may act as a protein kinase. Simply knowing that a protein adds phosphates does not reveal how it functions in a living organism. Additional information is required to understand the context in which the biochemical activity is used. What are its protein targets? In which tissues is it active? Which biological pathways does it influence? What role does it have in the growth or development of the organism?
This article will discuss methods to determine protein structure, which is certainly still important and no easy feat, as well as its function. A lot of the information I got in this article was from a conversation I had with Professor Lars Konermann at The University of Western Ontario, whos lab focused on protein folding, protein interactions with drugs and other molecules, and the mechanisms of protein aggregation (clumps of improperly folded or unfolded proteins that can form both inside and outside of cells). A lot of his work involves using experimental techniques to determine protein structure. Finally, another cool branch of his lab explores the mechanisms of electrospray ionization: the transition of proteins and other biological molecules from solution into the gas phase. We will discuss why that is important later.
Starting with the amino acid sequence of a protein, one can often predict which secondary structural elements, the two main ones being alpha helices and beta sheets, will be present in the protein. Yet, it is still a challenge to reliably deduce the three-dimensional folded structure of a protein from its amino acid sequence. The main techniques for a long time has been experimental methods such as X-ray crystallography and NMR spectroscopy, yet computational methods, particularly deep learning-based approaches like AlphaFold2, AlphaFold3 and RoseTTAFold (an extra paper on RoseTTAFold), have recently achieved remarkable success in predicting structures from sequences and much more — notably protein interactions, the structure of assemblies of proteins, and even designing molecules from scratch.
Experimental methods: x-ray crystallography, NMR spectroscopy, and cryo electrode microscopy.
While these computational methods are now revolutionary, they would not be possible without the initial, hard work from experimental methods such as X-ray crystallography. In short, to use this technique, the crystallographer obtains protein crystals, records the diffraction pattern formed by x-rays passed through the crystals, and then interprets the data using a computer. The result is a atomic-resolution model of a protein. Lets examine this in more detail
Proteins must be first crystallized in order to collect data on them. Protein crystallization involves transitioning proteins from a dissolved state in a solution into a solid, highly ordered, repeating structure. In a bit more detail, the concentrated protein solution is subjected to a wide variety of crystallization conditions: temperature, pressure, solvent type, solute concentration, cooling rate. Many different conditions are tried in parallel because we don’t know beforehand which conditions are needed for obtaining crystals for a given protein.
Proteins have hydrophilic and hydrophobic side chains. Water soluble proteins fold in a way that the hydrophobic ones (the water hiding ones) are hidden inside. The hydrophilic chains are on the outside which makes the protein pretty stable and easy to work with. They even crystalize quite well. Even if some proteins are soluble, such as membrane proteins, it can still be very hard to crystallize them because their structure is dependent on the membrane that they are attached to. However, if you want to look at a protein, you want to have just the protein in the sample, essentially purifying it. This conflict means simulating membrane proteins or just membrane themselves is an active area. Here is one particularly good example.
The Nobel Prize in 1988, was the first membrane protein to be crystallized ever. Ever since there has been slow and steady progress. Even computational models struggle to accurately model membrane proteins because they don’t understand that the protein is attached to the membrane and they also rely on evolutionary data, which is limited for membrane proteins. There is a pretty cool solution outlining crystallizing membrane proteins using lipidic mesophases. Here is another good overview of membrane protein solutions if you are interested.
X-rays, like light, are a form of electromagnetic radiation, but they have a much shorter wavelength, typically around 0.1 nanometers — the diameter of a hydrogen atom. If a narrow parallel beam of x-rays is directed at a sample of a crystal, most of the x-rays pass straight through it. A small fraction, however, interacts with the crystal and experience a change in some of there properties: position, amplitude, and phase. We call these changes “diffraction,” and all the diffracted x-rays are collected in a diffraction pattern. The crystal is then rotated sightly and a new diffraction pattern is obtained. This process is repeated through 360 degrees along one axis (typically rotations through a smaller angle on another axis are also recorded to avoid blind spots) until the instrument has recorded a diffraction pattern for each position. This makes sense, if you saw my hand at all of its orientations, you would confidentially be able to produce a 3D structure of it.
These changes by the diffracted x-rays obey a mathematical concept called the Fourier transform. To get a model of our protein structure, we feed back the diffracted x-ray data into the Fourier transform, and reciprocate it (raise it to the power of minus one).
To reciprocate the Fourier transform and deduce a a three-dimensional map of all the electron densities (where electrons are) in crystal — or an electron density map — we require the following information:
The relative position of the diffracted waves.
The relative amplitudes of the diffracted waves.
The relative phases of the diffracted waves.
We can directly measure the first two using an X-ray detector. All the diffracted X-rays hit the detector with a definite position and produce a signal proportional to their amplitude. But we lose all the phase information. Nor can we measure it directly or indirectly.
This is called the phase problem.
The two most common ways of overcoming the phase problem in protein crystallography are Molecular Replacement and SAD phasing.
Molecular Replacement
We can take the phases from another structure that resembles the one we are trying to solve. Combine them with the symmetry of the new crystal, and do the inverse Fourier transform. With some luck, we get an electron density map with interpretable features.
SAD (Single-wavelength Anomalous Dispersion) Phasing
It exploits two properties of diffraction patterns:
Every single atom contributes to every bit of the diffraction pattern.
Atoms with lots of electrons (”heavy atoms”) make the greatest contribution to the diffraction pattern.
These” heavy atoms” cause small, measurable differences in the diffraction pattern that depend on the phase, so we can deduce the missing phase information.
Once we have the electron density map. Since the electrons orbit atomic nuclei or exist between them in bonds, the electron density map is, roughly a ball-and-stick model of the protein structure.
Interpreting this map—translating its contours into a three-dimensional structure—is a complicated procedure that requires knowledge of the protein’s amino acid sequence of. Largely by trial and error, the sequence and the electron-density map are correlated by a computer to give the best possible fit.
What Information Do We Get from Protein Crystallography?
In a successful case, you get an atom-by-atom model of the protein structure and a set of statistics that tell you roughly how accurate it is. Depending on the quality of the data, you may get some bonus information, such as the water molecules that form part of the quaternary structure. Or you may be able to tell if a disulfide bond has formed or if an amino acid is modified.
You’ll usually be able to see if the protein is bound to a ligand, but accurately building the ligand into the model might be difficult. The granularity and information-richness of a structure largely depend on the resolution of the diffraction data.
Despite this great data, these static structures don’t tell you anything about folding or dynamics. Normally, proteins are not static in the body because they are constantly being pushed around by water molecules in our body. Knowing dynamics and folding is important because we need to know how it would interact with different molecules and how it will look like in the body when molecules are colliding and the structure is being slightly altered.
Nuclear Magnetic Resonance (NMR) Spectroscopy
NMR spectroscopy has been widely used for many years to analyze the structure of small molecules, and is increasingly applied to the study of small proteins. Unlike x-ray crystallography, NMR does not depend on having a crystalline sample; it simply requires a small volume of concentrated protein solution that is placed in a strong magnetic field. Thus, NMR is especially useful when a protein of interest has resisted attempts at crystallization, a common problem for many membrane proteins. Because NMR studies are performed in solution, this method also offers a convenient means of monitoring changes in protein structure, for example during protein folding or when a substrate binds to the protein. NMR is also used widely to investigate molecules other than proteins and is valuable, for example, as a method to determine the three-dimensional structures of RNA molecules and the complex carbohydrate side chains of glycoproteins.
NMR exploits the magnetic properties (particularly the property spin) of certain atomic nuclei, most often hydrogen-1 but also carbon-13 and nitrogen-15, to figure out how atoms are arranged in a molecule. In response to pulses of electromagnetic radiation, the spin of the atoms, which is normally aligned along the strong magnetic field, can be changed to a misaligned excited state.
When the excited nuclei return to their aligned state, they emit radiation, which can be measured and displayed as a spectrum. The nature of the emitted radiation depends on the environment of each hydrogen nucleus, such as the atom type (backbone vs side chain), neighboring atoms and bonds ( if one nucleus is excited, it influences the absorption and emission of radiation by other nuclei that lie close to it), and the secondary structure it is apart of (α-helix, β-sheet, loops). Because chemical shifts are sensitive to structure, they act like a "fingerprint" for each atom. If we know which shifts belong to which atoms, and we measure how spins interact (through bonds or through space), we can piece together:
Connectivity (which atoms are bonded)
Distances (which atoms are close in space)
Angles (bond angles from coupling constants)
Combining all this and the knowledge of the amino acid sequence makes it possible to produce the three-dimensional structure of the protein.
Cryo electrode microscopy (cryo-EM)
Cryo-EM combines the power of electron microscopes to see tiny things and the power of cryogenic temperatures to preserve their natural shapes and structures. (Think freezing, freezing cold, around −183 °C or nearly –300 °F: more than twice as cold as the coldest surface temperature ever recorded on Earth). Arguably the most exciting entry point to structural biology in recent years, today’s cryo-EM is known for the remarkable and ever-increasing precision with which it “solves” molecular structures, earning cryo-EM’s early pioneers the 2017 Nobel Prize in Chemistry.
Cryo-EM can capture flexible, wiggly proteins not suited to x-ray crystallography, and it can illuminate big molecules like intact viruses, giving it an advantage over NMR, which is limited to small molecules.
In summary, cryo-EM freezes many copies of a delicate sample into a glassy state, hits them with an electron beam to create numerous 2D shadows, and then, using some sort of software, groups these 2D shadows to create a 3D model of the sample.
Quantum mechanics tells us that fundamental bits of matter like the electron are both particles and waves. Cryo-EM takes advantage of the fact that the electron has a very tiny wavelength – much shorter than wavelengths of light – so it can make clear images of equally tiny things. However, an intense electron beam can damage or destroy a delicate sample. Thus, the samples are frozen to reduce the radiation damage caused by the electron beams. The freezing method also must be incredibly specific: doesn’t form ice crystals, which happens when water freezes slowly and would damage the delicate molecular structures, and that’s fast enough to capture that natural state.
Cryo-EM gets around both of those problems by freezing samples into a glass-like state.
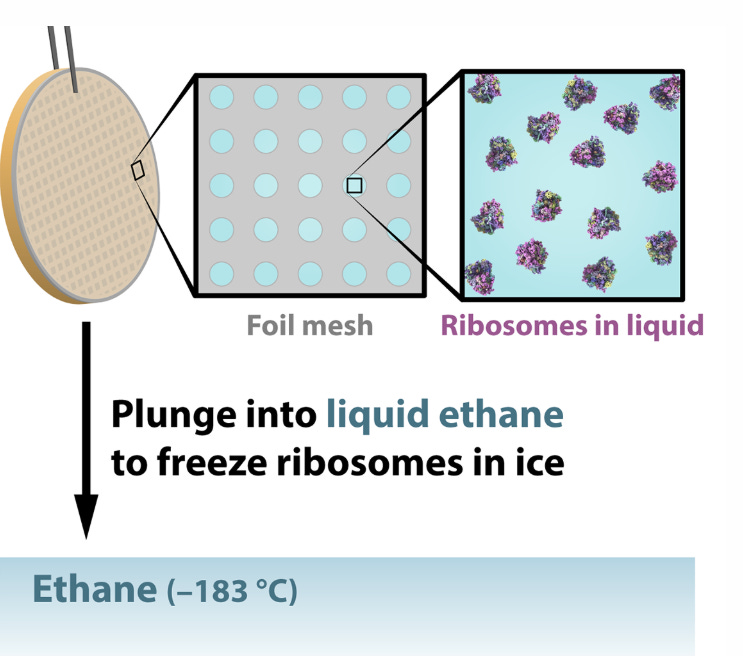
First, researchers extract the specimens they want to study – lets say a protein molecules in this case — and suspend them in water, where they float freely. Next, they deposit a drop of water containing thousands of copies of the protein across a grid of tiny holes in a carbon net.
A robotic arm plunges the grid into liquid ethane that’s been cooled in a bath of liquid nitrogen, and the water and its cargo of proteins instantly freeze into a glassy state.
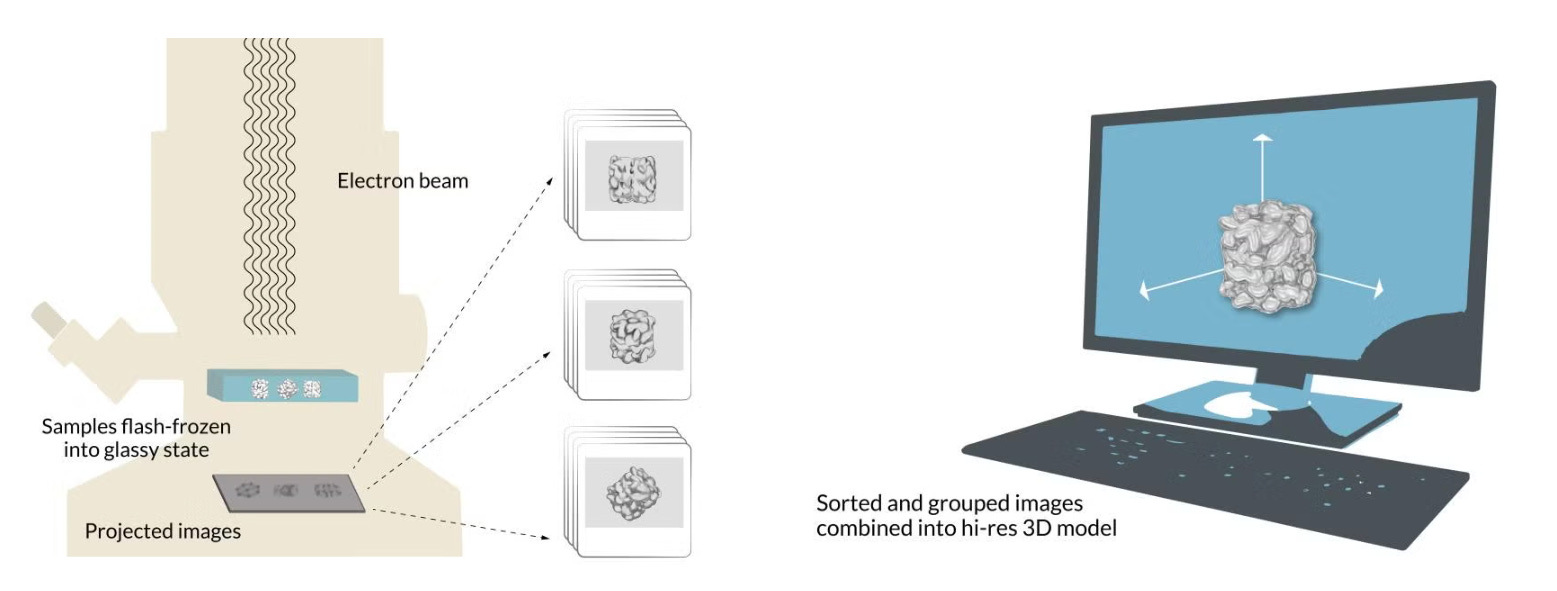
When the sample is flash frozen, the proteins trapped in the ice are ideally arranged in random orientations, so that we’ll be able to image their structures from all angles. Placed into a transmission electron microscope and shot with an electron beam, these random orientations appear as 2D shadows on a detector, producing many thousands of images of different views of the particles.
Because the intensity of the electron beam is set as low as possible to limit the inevitable structural damage to the sample, all of the images we have now collected appear grainy. This means we have a considerable amount of “noisy” images — images with unwanted background “noise” relative to the meaningful “signal” of the image.
To improve this “signal-to-noise ratio,” we pick out the best 2D images of our proteins before computer software groups them together by orientation (up, down, sideways, and so on). We can now build a representative image — an average of all the 2D images at said orientation (a 2D class average) — for every orientation that has a much improved signal-to-noise ratio.

{kind=link}
This array of 2D images is combined again into a 3D reconstruction of the molecule. Researchers can move and rotate this reconstruction in a computer to look at the protein from all sides. If you want to learn more about cryo-em, check out these two cool papers: 1, 2.
Cryogenic electron tomography (cryo-ET)
The cryo-EM technolgy is of course evolving, and one cool example is a development of a form of cryo-EM called called cryogenic electron tomography. Cryo-ET is the only structural biology method available today that can image everything from molecules to cells to tissues to small organisms in their near-native context. Whereas cryo-EM takes many frames of many particles on the grid, cryo-ET takes many frames of the same sample as it is tilted under the microscope to capture different parts of its structure. Computational steps align and merge these images, forming a tomogram, or a 3D picture that can illuminate how molecules carry out their work inside the cell.
Yet, solving structures using experimental methods requires extremely specialized training, a high degree of skill, and a lot of luck. The cost of solving a new, unique structure is on the order of $100,000. Given the difficulty of solving an experimental structure, and considering the rate at which new protein sequences are discovered, it has become clear that with today’s technology, we will not solve structures for all the new proteins being identified and sequenced.
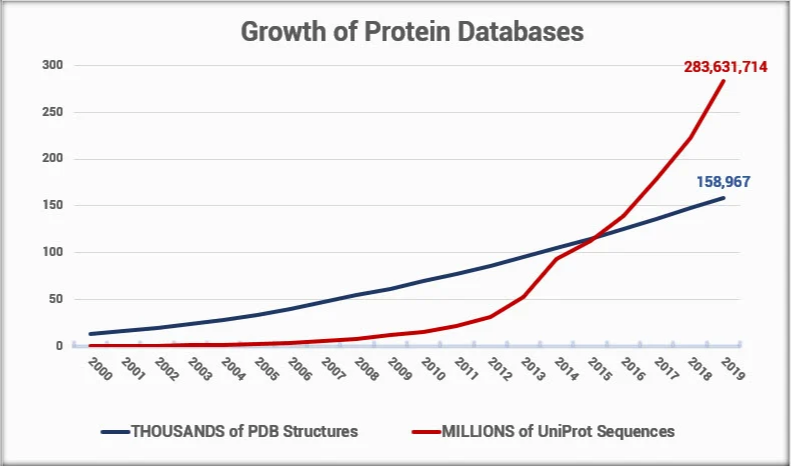
Finding alternative ways to predict a protein structure becomes more and more important as this gap
Sequence Similarity Can Provide Clues About Protein Function
Because amino acid sequence determines protein structure and structure dictates biochemical function, proteins that share a similar amino acid sequence usually perform similar biochemical functions, even when they are found in distantly related organisms. Since there is ample data on protein and nucleic acid sequences, the function of a gene—and its encoded protein—can often be predicted by simply comparing its sequence with those of previously characterized genes. At present, determining what a newly discovered protein does therefore usually begins with a search for previously identified proteins that are similar in their amino acid sequences. This is in fact one of the first steps AlphaFold does. It compares and aligns multiple proteins, DNA, or RNA that have similar sequences with the target protein’s — the one with the 3D structure we want to predict — sequence through a process called MSA (multiple sequence alignment). Predictions that emerge from sequence analysis are often only a tool to direct further experimental investigations. It is definitely not the determining step for a proteins function.
Fusion proteins to analyze protein function and track proteins in living cells
The location of a protein within the cell often suggests something about its function. Proteins that travel from the cytoplasm to the nucleus when a cell is exposed to a growth factor (literally just something that can stimulate a cell to grow or proliferate), for example, may have a role in regulating gene expression — the process where a gene's instructions are used to create a functional product, usually a protein — in response to that factor.
A protein often contains short amino acid sequences that determine its location in a cell. Most nuclear proteins, for example, contain one or more specific short sequences of amino acids that serve as signals for their import into the nucleus after their synthesis in the cytosol. These special regions of the protein can be identified by fusing them to an easily detectable protein that lacks such regions and then following the behavior of this protein in a cell.
Another common strategy used both to follow proteins in cells and to purify them rapidly is epitope tagging. In this case, a fusion protein is produced that contains the entire protein being analyzed plus a short peptide of 8 to 12 amino acids (an “epitope”) that can be recognized by a commercially available antibody. The fusion protein can therefore be specifically detected, even in the presence of a large excess of the normal protein, using the anti-epitope antibody and a labeled secondary antibody that can be monitored by light or electron microscopy.
Large numbers of proteins are also being tracked in living cells by using a fluorescent marker called green fluorescent protein (GFP). Tagging proteins with GFP is as simple as attaching the gene for GFP to one end of the gene that encodes for a protein of interest. In most cases, the resulting GFP fusion protein behaves in the same way as the original protein, and its movement can be monitored by following its fluorescence inside the cell by fluorescence microscopy.
GFP and its color variants can be used to see protein-protein interactions. The method works by attaching a different glowing tag to each protein. These tags are chosen so that the light given off by the first tag can be absorbed by the second one. If the proteins come very close together—within a few nanometers—the energy from the first tag transfers to the second. This process is called fluorescence resonance energy transfer (FRET). Scientists detect this by shining light on the first tag and checking if the second one glows. Using different versions of GFP, researchers can monitor the interactions of any two protein molecules inside a living cell.
Protein affinity chromatography and co-immunoprecipitation allow the identification of associated proteins
Because most proteins in the cell function as part of a complex with other proteins, an important way to begin to characterize their biological roles is to identify their binding partners. If an uncharacterized protein binds to a protein whose role in the cell is understood, its function is likely to be related. For example, if a protein is found to be part of the proteasome complex, it is likely to be involved somehow in degrading damaged or misfolded proteins.
Protein affinity chromatography is one method that can be used to isolate and identify proteins that interact physically. To capture interacting proteins, a target protein is attached to polymer beads that are packed into a column. When a mixture of cellular proteins is passed through, only the ones that naturally bind to the target protein will stick to the beads; the rest wash away. Later, the bound proteins can be released (eluted) from the beads and identified using tools like mass spectrometry.
Perhaps the simplest method for identifying proteins that bind to one another tightly is co-immunoprecipitation. In this approach, an antibody is used to grab a specific protein. The antibody is attached to a solid support so it can pull the protein—and anything stuck to it—out of a solution. If another protein is tightly bound to the target protein, it will come along too. This technique helps researchers identify groups of proteins that work together inside cells, even if their interactions are brief, such as when cells respond to signals.
Co-immunoprecipitation techniques require having a highly specific antibody against a known cellular protein target, which is not always available. One way to overcome this requirement is to use recombinant DNA techniques to add an epitope tag or to fuse the target protein to a well-characterized marker protein, such as GST (glutathione S-transferase). Because antibodies for these tags and markers are easy to get, researchers can use them to pull out the tagged protein along with any partners it’s bound to. In the case of GST, you may not even need antibodies—GST naturally sticks to glutathione, so the fusion protein and its partners can be captured using beads coated with glutathione.
Methods such as co-immunoprecipitation and affinity chromatography allow the physical isolation of interacting proteins. A successful isolation yields a protein whose identity must then be ascertained by mass spectrometry, and whose gene must be retrieved and cloned before further studies characterizing its activity—or the nature of the protein-protein interaction—can be performed.
Other techniques allow the simultaneous isolation of interacting proteins along with the genes that encode them. Two-hybrid screening uses a reporter gene to detect the physical interaction of a pair of proteins inside yeast cells. The system is designed so that when two proteins interact, they bring together two separate halves of a gene activator. This activator switches on a “reporter gene,” signaling that the two proteins have bound to each other.
This method works because gene activator proteins are modular—they usually have one part that binds DNA and another part that turns on transcription. Scientists take advantage of this by attaching the DNA-binding half to a “bait” protein (the protein they want to study). This bait protein then anchors at the control region of a reporter gene in the yeast nucleus.
To search for partners, scientists create a library of potential “prey” proteins by linking random DNA fragments to the activation half of the activator protein. Each yeast cell is given a different prey construct. If the prey protein binds to the bait, the DNA-binding half and activation half are brought together, turning on the reporter gene. The yeast cells that light up this reporter are then isolated, and researchers can sequence the DNA to identify which prey protein was interacting with the bait.
If this process sounded confusing imagine you’re trying to figure out which pairs of puzzle pieces fit together. But instead of looking at the shapes, you set up a lamp that only turns on when the right two pieces snap together.
The bait protein is like the first puzzle piece you already know and place into the lamp system.
The prey proteins are all the other puzzle pieces you want to test.
If a prey piece clicks perfectly with the bait, the lamp lights up — just like the reporter gene switching on.
By seeing which puzzle piece made the lamp turn on, you know which protein interacts with your bait, and you can then look up the “label” (the gene) for that puzzle piece.
Two-hybrid screening is relatively simple to use in the laboratory. Although the protein-protein interactions occur in the nucleus of yeast cels, proteins from every part of the cell and from any organism can be studied in this way.
A variation called the reverse two-hybrid system is used to find mutations or chemicals that block two proteins from binding. Instead of using a reporter gene, this system uses a gene that actually kills the yeast cell if the bait and prey proteins bind. That way, only cells where the proteins fail to interact — because an engineered mutation or a test compound prevents them from doing so — will survive. Eliminating a particular molecular interaction can reveal something about the role of the participating proteins in the cell. In addition, compounds that selectively interrupt protein interactions can be medically useful: a drug that prevents a virus from binding to its receptor protein on human cells could help people to avoid infections, for example.
Another method for finding protein–protein interactions uses viruses that infect the bacterium E. coli (called bacteriophages, or “phages”). In this approach, the DNA encodingor the protein of interest—or a smaller peptide fragment of this protein—is fused with a gene encoding one of the proteins that forms the viral coat. When the virus infects E. coli it replicates, producing new viral particles that display the hybrid protein on their outer surface. These phages, carrying the protein of interest on their coats, can then be used to search through a large collection of proteins to identify binding partners.
The most powerful use of phage display is to screen huge collections of proteins or peptides to see which ones bind to a chosen target. To do this, scientists first create a library of phages, each carrying a different fusion protein—similar to the prey library used in the two-hybrid system. This phage library is then tested against a purified target protein. For example, the phages can be passed through a column that contains the target protein fixed in place. Any phages displaying a protein or peptide that sticks tightly to the target will stay behind in the column, while the rest wash away. These bound phages can then be released (eluted) by adding extra target protein.
The selected phages, which carry the DNA encoding the interacting protein or peptide, are collected and allowed to multiply inside E. coli. Researchers then recover the DNA from these phages and determine its nucleotide sequence to identify the binding protein or peptide. A similar version of this method has been used to find peptides that specifically attach to the inner lining of blood vessels in human tumors. These peptides are now being tested as a way to deliver anti-cancer drugs directly to tumors.
DNA footprinting reveals the sites where proteins bind on a DNA molecule
So far we have concentrated on examining protein-protein interactions. But some proteins act by binding to DNA. Most of these proteins, by binding to regulatory DNA sequences usually located outside the coding regions (regions which encode for RNA) of a gene, have a central role in determining which genes are active in a particular cell.
In analyzing how such a protein functions, it is important to identify the specific nucleotide sequences to which it binds. A method used for this purpose is called DNA footprinting. First, a pure DNA fragment is prepared and labeled at one end with radioactive phosphorus (32P). This DNA is then cut randomly at single sites using either a nuclease enzyme or a chemical that makes single-stranded breaks. After heating (denaturing) the DNA to separate its two strands, the labeled strand fragments are separated on a gel and visualized by autoradiography.
The key step is comparing two samples: one with a DNA-binding protein present and one without. If the protein is bound, it shields the nucleotides at its binding site and prevents those bonds from being cut. As a result, the DNA fragments that would normally end within the binding site are missing from the gel. This absence shows up as a clear gap in the banding pattern, called a “footprint.” Similar approaches can also be applied to map where proteins bind on RNA.
Computational Methods
Computational methods, while not perfect, have been revolutionary in quickening our pace of determining protein structures and uncovering use cases for proteins. While they won’t ever replace experimental methods, that doesn’t take away from their importance.
AlphaFold models are already useful for many practical aspects of structural biology: to design better protein expression experiments; to solve experimental structures faster, especially for X‐ray crystallography; to overcome tedious model building steps in experimental crystallographic and cryo‐EM maps; and to interpret lower‐resolution cryo‐EM maps.
Above all, the analysis of the models themselves can generate new and testable hypotheses about protein function. This actually is the great joy of structural biology: to derive mechanistic insight from a protein structure. In a sense, AI provides structural biologists with a new technique, bringing the fun of structure‐gazing without the effort of experimental work.
To better understand the enormous advance that AlphaFold and RoseTTAfold have achieved, it is worthwhile to look at the problem. Protein folding involves rearranging a linear sequence of amino acids in space to a physiologically preferred low‐energy state. Predicting the correct structure from the amino acid sequence alone and determining how the amino acid sequence dictates this 3D structure was deemed an intractable problem. The degrees of freedom in the peptide bonds create an astronomically high number of possible structures: going through all the possibilities would take longer than the age of the universe, even for a small protein (Levinthal’s Paradox).
The “protein folding problem” consists of three closely related puzzles:
What is the folding code?
What is the folding mechanism?
Can we predict the structure of a protein from its amino acid sequence?
Computational predictions were developed to circumvent testing protein structures one by one (sequential sampling). Over the past 40 years, these methods have steadily improved by comparing protein sequences to the growing number of protein structures solved through the aforementioned experimental methods — all freely available in the Protein Data Bank (PDB)
In 1994, the scientific community began a bi-annual competition (CASP) to test how well these computational tools could predict protein structures. The challenge was to predict the shape of proteins that had just been experimentally solved but not yet released to the public. Over the decades, gradual improvements were made, but everything changed with the arrival of AlphaFold. Its first version, launched only three years ago, applied artificial intelligence to the problem and was already a major breakthrough. By 2020, a redesigned version of AlphaFold achieved near-perfect predictions, creating a seismic shift not only in structural biology but also across many fields of science.
I already wrote an article outlining how AlphaFold works but “in short” (I enjoy discussing this stuff):
The user inputs a protein sequence. The principle used in protein folding models is that proteins with similar functions often have similar structures and thus sequences. AlphaFold2 thus aligns this sequence with multiple similar protein sequences and builds a multiple sequence alignment (MSA).
Sequences from various species are aligned in a 2D table format, placing corresponding amino aid residues — the parts of the amino acid that remain after two or more amino acids combine to form a peptide — in the same column and species in different rows. Most known protein sequences (about 70%) have at least one part that’s similar to a protein with a known structure. This is why MSA is crucial for predicting protein structure.
A high-quality MSA is essential for AlphaFold2 to produce an accurate prediction of protein structure. A diverse and deep MSA, with hundreds or thousands of sequences in the alignment, will help AlphaFold2 to identify co-evolutionary signals, conserved regions (parts that stay the same across different species), understand how mutations might affect the protein, and use all of these clues to accurately predict the protein’s 3D structure. A shallow MSA, with only tens of sequences and low variability among them will struggle to identify these patterns and is the most common reason for failing, non-confident and inaccurate AlphaFold2 predictions. Co-evolution, or covariation, is a process that allows the protein to preserve its structure even as it evolves. If two amino acids are in close contact, a mutation in one will be followed by a mutation in the other. Conversely, if two regions of a protein are changing and evolving independently from each other, it is likely that they are not in direct contact
AlphaFold2 also uses the MSA to predict and generate a set of pair representations modeling the interactions between every pair of amino acid residues based on the secondary structure of the protein, regardless of distance.
The MSA and pair representations are passed through AlphaFold2’s neural network (Evoformer) to interpret and refine both the MSA and the pair representations. AlphaFold2’s structure module takes both the refined pair representation and the original sequence (from the MSA) from the Evoformer. The structure module first turns this into a backbone of the 3D structure. It then finishes the modeling by placing the amino acid side chains and refining their positions.
If available, AlphaFold2 can feed supplied protein structures (e.g. structures derived from experiment) as templates into the Evoformer. However, AlphaFold2 tends to ignore such templates if there is enough information coming from the MSA. It is significant that AlphaFold2 doesn’t necessarily need templates as this differentiates it from homology modeling. Homology modeling is a computational method for predicting the three-dimensional structure of a protein based on its amino acid sequence and the known structure of a homologous protein (a protein with a shared evolutionary ancestry). It leverages the principle that proteins with similar sequences often share similar structures. AlphaFold2 doesn’t necessarily need these homologous proteins (templates) as data points for its structure prediction, meaning it can predict protein structures with a more limited evolutionary history — though AlphaFold2 still relies on evolutionary data through the MSA even if it doesn’t require homologous proteins.
AlphaFold2, and this step is incredibly important, then performs an iterative process called “recycling”. It feeds the MSA, the pair representations and the 3D structure back to the neural network, and generates a new 3D structure. This process is repeated numerous times, allowing AlphaFold2 to improve the accuracy of the final structure.
Along with the 3D structure, AlphaFold also provides confidence metrics, such as pLDDT, pTM and PAE enabling critical interpretation.
Essentially, AlphaFold2 analyzes 1-D data and based on that, predicts 2-D interactions. Both are refined and used to update each other, and then are finally used to predict the 3-D structure. The 1-,2-, and 3-D data is then all passed back to generate a new 3-D structure until no more improvements can be made.
RoseTTAFold, while not as accurate as AlphaFold2, uses significantly less computational energy and can thus predict the structures of large proteins on a gaming laptop. RoseTTAFold has a very similar architecture to AlphaFold and similarly relies on MSA. I won’t delve into how it specifically works but here is a great article if you want to learn more.
Gaseous protein folding
Professor Lars Konermann and I discussed another aspect of his lab and that is gaseous protein folding. In water, the driving force for protein folding is hiding the hydrophobic chains: it is the fear of water that drives things together.
In a vacuum, because there is no water, the opposite happens. The hydrophobic chains are very happy on the surface. The things that normally interact with water, the positively and negatively charged side-chains, would rather be packed together on the inside. Thus, in the absence of water, the hydrophobic side chains get squeezed to the outside and you get these super cool “inside out” structures. What is driving folding in a gaseous environment is the hydrophilic chains wanting to interact with something and so they pack together.
This is important because when mass spectrometry experiments happen, they occur in a vacuum or in a gaseous state, so its interesting and important to explore how the proteins behave in these areas. Since we bring a protein from a solution to the gas phase when we want to study its structure, the chemical reaction — the conversion of a properly folded protein into an inside-out protein — is incredibly slow because there is a high activation barrier. An activation barrier (or activation energy) is the minimum amount of energy that molecules need to overcome in order for a reaction or structural change to happen. To get the protein to rearrange itself into a new conformation (e.g., “inside-out”), you would need to supply a lot of extra energy — through heat, collisions, or some external force. If that energy isn’t supplied, the protein will basically stay as it is, because it’s energetically unfavorable to cross the barrier. This means that if you bring a properly folded protein into the gas phase, and if you do it under gentle conditions, you can actually preserve its structure very well because it requires an insane amount of energy to overcome the activation barrier again to unfold the protein.
You can also just start with the unfolded state of the protein and add heat to give it enough energy to overcome the activation barrier and properly fold. This is because when you add heat, you’re increasing the amount of energy each atom/molecule has in its vibrational, rotational, and translational motions.
Another application of gaseous protein folding is for drug development. Whenever you develop drugs, the first step in a drug action mechanism is always binding. If you have a small molecule and it does something to your body, that's always because it binds to something. In most cases, it will bind to a protein. For example, penicillin works by binding and blocking an enzyme’s active site.
To find a molecule that nicely binds to a protein is actually very difficult because its difficult to measure how effectively one molecule binds to another in a solution. However, if you bring a protein from solution into the gas phase, all you have to do is measure mass: if the mass increases it means that the molecule successfully bound to something.
You can do this in the pharmaceutical industry where you mix the protein of interest with thousands of compounds at a time and you can see exactly which one binded by measuring the weight of this new molecule. Mass is actually a very unique identifier in chemistry, so if you know the mass of each small molecule and the protein, the resulting mass of the compound would tell you if a molecule successfully binded and, if so, which one.
Thank you so much for taking the time to read this article! If you have any feedback, feel free to let me know!