
mRNA Vaccine Technology: Advantages, Limitations, and Future Trajectory
A history of vaccine technologies and the trajectory of mRNA vaccines.

This post may not be for everyone; it consists of a paper I wrote about mRNA vaccines. This was my first time writing a scientific paper on my own and so the jargon may be too technical and convoluted for most to understand. But if you want an in-depth history of vaccine technologies and mRNA vaccines, and are willing to put up with my crazy writing, then please take a read and let me know what you think.
Abstract
Vaccines are one of the most impactful biomedical developments in history. They prevent between 3.5–5 million deaths every year and have helped eradicate diseases such as smallpox and polio. Recently, vaccines have significantly reduced the severity of symptoms and mortality caused by SARS-CoV-2. Despite incredible advances in vaccine technology, including the most recent development and commercialization of messenger RNA (mRNA) based vaccines for SARS-Cov-2, there remain several limitations. Vaccine induced immunity is often limited in its breadth, resulting in poor protection to rapidly mutating viruses, and its durability, requiring patients to receive a complex booster regime. Furthermore, many vaccines require complex storage conditions relying on the cold chain, preventing equitable global access in regions that lack the infrastructure. This review will discuss recent advances in vaccine technologies and highlight mRNA vaccine strategies that seek to overcome current technological challenges to create next generation vaccines that are more biomimetic, durable, and potent.
Keywords
Vaccines, mRNA, Immunology, Lipid nanoparticles, Vaccine technology
Introduction
Vaccines are a biological product intended to safely stimulate and prepare the immune system against infection or disease. They leverage the ability of the mammalian innate and adaptive immune systems to recognize, respond to, and remember pathogens, and in countries with high vaccination rates, many diseases responsible for high childhood mortality have essentially disappeared. These successes notwithstanding, the recent COVID-19 pandemic demonstrated that infectious diseases with high incidence or fatality rates pose a substantial threat to both public health and the global economy. Thus, it has become imperative to be able to respond to such threats more rapidly. While strategies such as social distancing, quarantine, and lockdown control the spread of an emerging pathogen, they are logistically challenging and hard to enforce long-term. Globally accessible and potent vaccination is ultimately the best approach to limit disease transmission and establish herd immunity.
The earliest documented practice of variolation – intentionally exposing healthy people to a virus – took place in the 16th century when people attempted to prevent illness by exposing healthy people to smallpox pus or scabs (1). In 1774, Benjamin Jesty made a breakthrough, testing his hypothesis that infection with cowpox could protect a person from smallpox. Shortly after, in 1796, Dr. Edward Jenner observed that the milkmaids who had cowpox lesions were immune against smallpox infection (1-2). Jenner’s following work evaluating immune protection in James Phipps and other children laid the foundation for modern vaccinology, a term coined from the Latin word vacca for cow (3).
Over the following two centuries, advancements in biomedical technology and immunology enabled the design of live-attenuated, whole-inactivated, sub-unit, and mRNA vaccines, among other platforms. For example, the evolution of cell culture led to the creation of the polio vaccine, and soon afterward vaccines for measles, mumps, rubella, and varicella were developed (4). The introduction of recombinant DNA and whole-genome sequencing techniques were major milestones in vaccine development, giving researchers the tools to develop new vaccines against pathogens, something not possible before (1). Other important milestones in vaccine research are the development of recombinant viral vector vaccines (5), virus-like particle vaccines (6), protein-based vaccines (7), and toxoid vaccines (5-8). Most recently, an important milestone for next-generation vaccines was the development of mRNA vaccines, which garnered increased attention after their rapid development and approval for the COVID-19 pandemic. The facile scalability and expedited approval can be attributed to mRNA vaccines’ unique safety profile, ease of fabrication, and ability to harness the patient’s own cellular machinery to express the desired vaccine antigen, the protein or peptide against which an immune response should be generated.
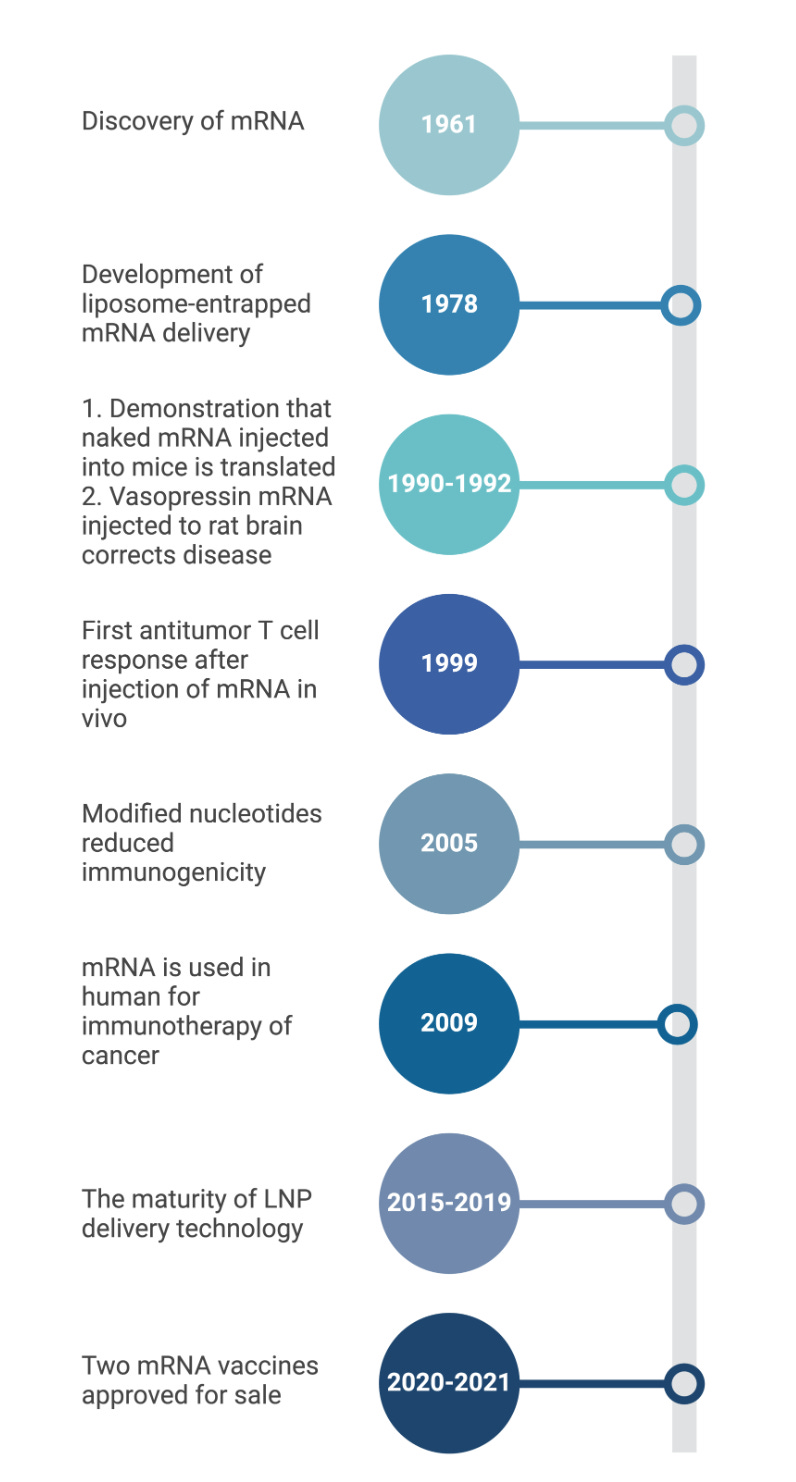
We are currently in the era of mRNA vaccinations. mRNA, which was discovered during pioneering studies between 1947–1961 (9), is a transient intermediate between genes and proteins (9). Efforts in the early 1990s showed that in vitro transcribed (IVT) mRNA vaccines can induce the production of proteins in animal models, with epitope presentation proving effective (10-11). In vitro transcription is a reaction in which a linearized DNA plasmid containing the gene of interest is transcribed to the mRNA sequence. Since the proof-of-concept animal studies in the early 1990s, numerous strategies have been explored to ameliorate the instability and immunogenicity of IVT mRNA (12). The true importance and efficacy of this vaccine technology, however, was realized when mRNA vaccines were developed and approved for the COVID-19 pandemic. They were developed in a record-breaking time of less than a year and their widespread vaccination to millions of people helped to control the COVID-19 outbreak. The development, approval, and manufacturing capabilities demonstrated by the makers of these vaccines has validated the mRNA platform as a safe and effective tool for vaccination. The goal of this review is to provide an overview of mRNA vaccines, focusing on their pharmacology, types, the unique immune responses they generate, mechanisms, benefits, and drawbacks.
Overview of Immune Response to mRNA vaccines
Vaccination is intended to mimic real infection as closely as possible without exposing the vaccine recipient to undue risks. There are two main mechanisms of the immune response: humoral and cellular immunity. In humoral immunity, B-lymphocytes cells (B cells) produce antibodies that bind to antigens, neutralizing them or preventing foreign substances (i.e viruses, bacteria, etc) from entering or damaging cells. Cellular immunity, on the other hand, relies on the activation of phagocytes, such as macrophages and natural killer cells, antigen-specific cytotoxic T-lymphocytes (CD8+ T cells), and the release of various cytokines in response to an antigen.
After a vaccine is administered, antigens are processed by a diverse population of immune cells known as antigen-presenting cells (APCs), which includes dendritic cells, macrophages, Langerhans cells, and B cells. Traditional vaccines, such as subunit or live attenuated vaccines, deliver whole proteins or peptides as their antigens while mRNA vaccines introduce a piece of mRNA that corresponds to a viral protein, acting as its antigen. The mechanism of antigen processing has important consequences on how immunity is induced. Vaccine antigens that are produced in or enter the cytoplasm (eg. live attenuated viruses) are displayed on class I major histocompatibility complexes (MHC-I) via the endogenous antigen-processing pathway (13). These antigens are then recognized by T cell receptors (TCR) of a particular subset: cytotoxic T cells (14). On the other hand, antigens that enter cells via phagocytosis are displayed on class II MHC (MHC-II) by the exogenous antigen-processing pathway (13). These antigens are recognized by a different subset of T lymphocytes: a naïve form of T helper cells (CD4+ T cells) (15).
mRNA vaccination, once administered intramuscularly, leads to potential adaptive immune system activation by the following pathways: (i) transfection of muscle and skin cells, activating immune cells and helping prime CD8+ T cells (16); (ii) transfection of tissue-resident immune cells such as the dendritic cells (DCs), macrophages, and Langerhans cells at the injection site, initiating priming and activation of not only T cells but also B cells (17); and (iii) transport to secondary lymphoid tissues, such as the lymph nodes and the spleen (17-18). After leveraging the translational machinery, such as the ribosome, of the host cell, the mRNA introduced by the vaccines is translated into proteins. The resulting translated proteins are processed and presented on either MHC-I or II. mRNA vaccines can then leverage both the endogenous and exogenous antigen-processing pathways. The endogenous antigen-processing pathway is used after proteasomes degrade cytoplasmic proteins, thus generating antigenic peptide epitopes that are loaded onto MHC-I molecules. MHC class I then can present these peptides on the surface of the cell for CD8+ T cells (19). This helps in establishing cellular immunity to the antigen expressed from the mRNA. Alternatively, the activation of APCs can result in the exogenous antigen-processing pathway, where secreted exogenous proteins are taken up by APCs, either residing in the tissue or draining lymph nodes, and then are processed and presented on MHC-II molecules (19).
The antigen is co-presented with macromolecular structures associated with pathogens. These pathogen associated molecular patterns (PAMPs) are recognized by pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) (20-21). The inclusion and design of these immunostimulatory molecules, often referred to as adjuvants, have become an active area of research, which will be more thoroughly considered in the following section. Briefly, the interaction between these adjuvants and PRRs on APCs triggers key intracellular signaling events that promote phagocytosis, maturation, and secretion of cytokines (22).
Activated APCs displaying vaccine antigens on MHCs then migrate to secondary lymphoid organs (eg., draining lymph nodes or spleen). Here APCs encounter naïve T cells, T cells that have not undergone activation, in T cell zones (23-24). The interaction between APCs and naïve T cells through MHC-TCR binding leads to the differentiation and proliferation of naïve T cells into effector cells.
In response to MHC-TCR binding, and cues from cytokines, CD4+ T-helper cells differentiate into two subsets of effector T-helper cells: T helper 1 (Th1) and T helper 2 (Th2) cells. Th1 and Th2 cells are primarily responsible for cellular and humoral immunity respectively. Th1 cells produce Interferon-gamma (IFN-γ) as their cytokine in order to stimulate the activation and expansion of CD8+ T cells. Th1 cells are further involved in the production of IgG1 and IgG3 antibodies by B cells (15). CD8+ T cells differentiate into cytotoxic killer T cells following TCR/MHC-I interaction and help from Th1 cells (e.g., INF-γ). Development of cytotoxic killer T cells following vaccination is important because they can recognize and eliminate infected cells. In addition to the effector cells that are generated in response to the presentation and recognition of vaccine protein antigens, both CD4+ and CD8+ T cells also differentiate into memory cells, which are critical in responding and expanding the clonal pool upon subsequent encounter with the same pathogen.
In promoting humoral immunity, Th2 cells secrete IL-4, IL-5, and IL-13 as their signature cytokines to promote the development, maturation, and differentiation of B cells into memory B cells (MBCs) and antibody-secreting plasma cells (Figure 1).
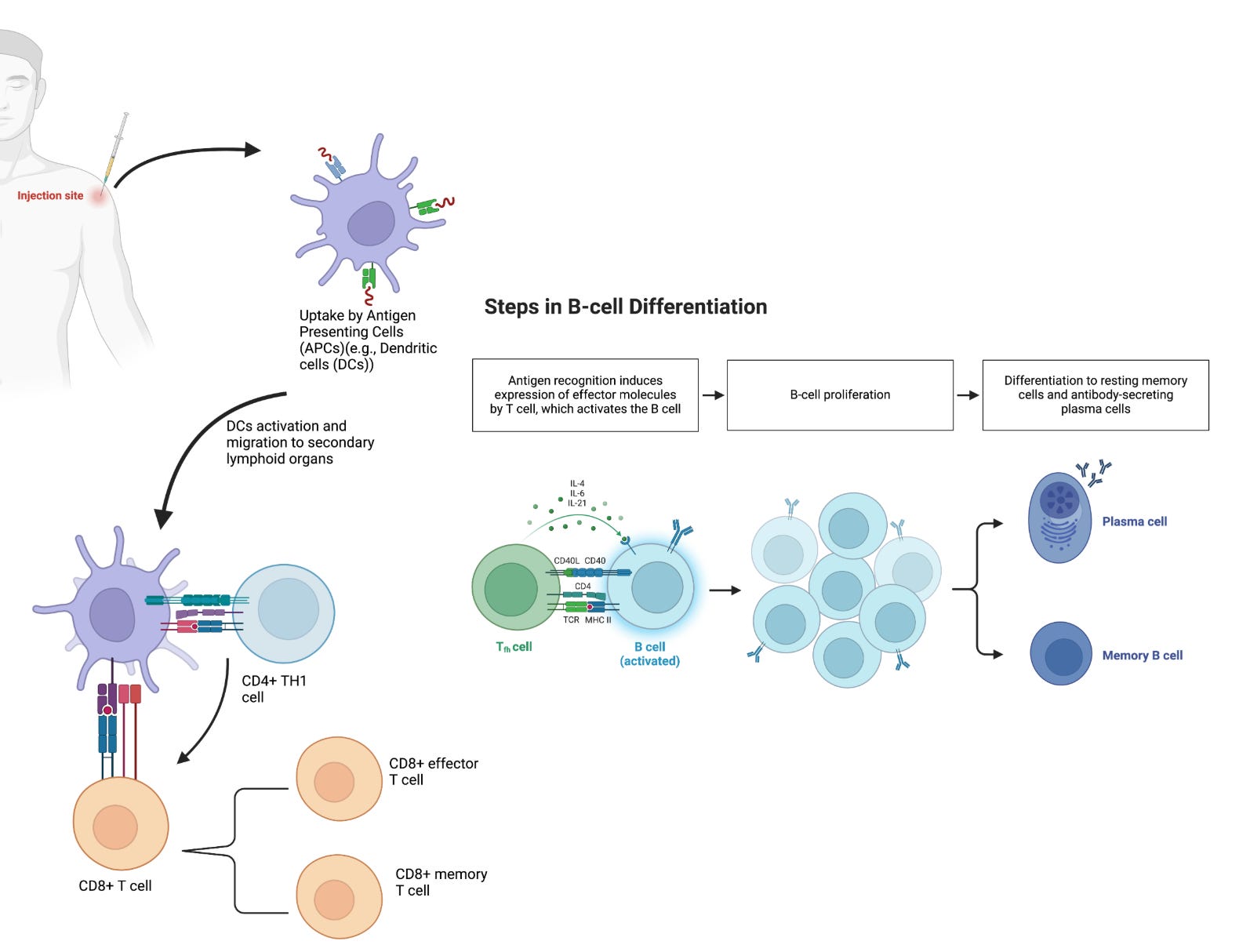
A number of additional T cell subclasses exist, each of which have niche roles and interactions in mediating immune responses to vaccines. For example, T follicular helper cells (Tfh) and Th17 cells are two additional subtypes of CD4+ cells that are essential for the generation of high-affinity antibodies and mucosal immunity. Tfh cells regulate B cell affinity maturation (somatic hypermutation), selection of high-affinity germinal center (GC) B cells, and the duration of GC reactions (14)(25). The quality of these processes ultimately determines the durability and potency of the humoral immune response through the generation of antibodies (by B cells) with high affinity. Antibodies are large (150 kDA MW) proteins that uniquely bind to epitopes on the antigens against which a patient is vaccinated. Having high quantities of circulating antibodies, or a robust memory response which can quickly generate new antibodies, is essential to preventing infection upon natural viral exposure. These circulating or memory-produced antibodies are capable of opsonizing viral particles, thereby preventing viruses from entering and infecting cells, or binding to secreted proteins, resulting in their degradation.
Durable GC reactions favor the differentiation of GC B cells into high-affinity MBCs and antibody-secreting long-lived plasma cells (LLPCs) (26)(27). MBCs are important in vaccine-induced immunity and for providing protection, as they can rapidly expand, and differentiate into short-lived antibody-secreting plasma cells upon antigen re-encounter (28). LLPCs migrate from the draining lymph nodes to the bone marrow, where they continue to produce antibodies for several months to decades. LLPCs are terminally differentiated, and, unlike MBCs, do not need reactivation or re-exposure to antigens. The high levels of neutralizing antibodies produced by LLPCs provide protection against reinfection.
B cells are also able to recognize and respond to vaccine antigens before T cells are engaged. Following vaccine administration, B cells detect and internalize antigens, and upon PRR activation, differentiate into plasmablasts, short-lived antibody-secreting cells, that produce the initial wave of antibodies.
The above aspects of the immune response help guide the development of vaccines and vaccine technologies to meet their intended use. For instance, the development of vaccines against intracellular infectious agents like viruses requires technologies or platforms that promote the endogenous antigen processing pathway or cross-presentation to induce potent cytotoxic T cell responses, which are essential for eliminating intracellular pathogens. Additionally, identifying vaccine technologies and/or adjuvants that effectively promote Tfh cell and GC responses, as well as generation of LLPCs, is vital for developing effective vaccines against both current and emerging infectious diseases.
Vaccine design
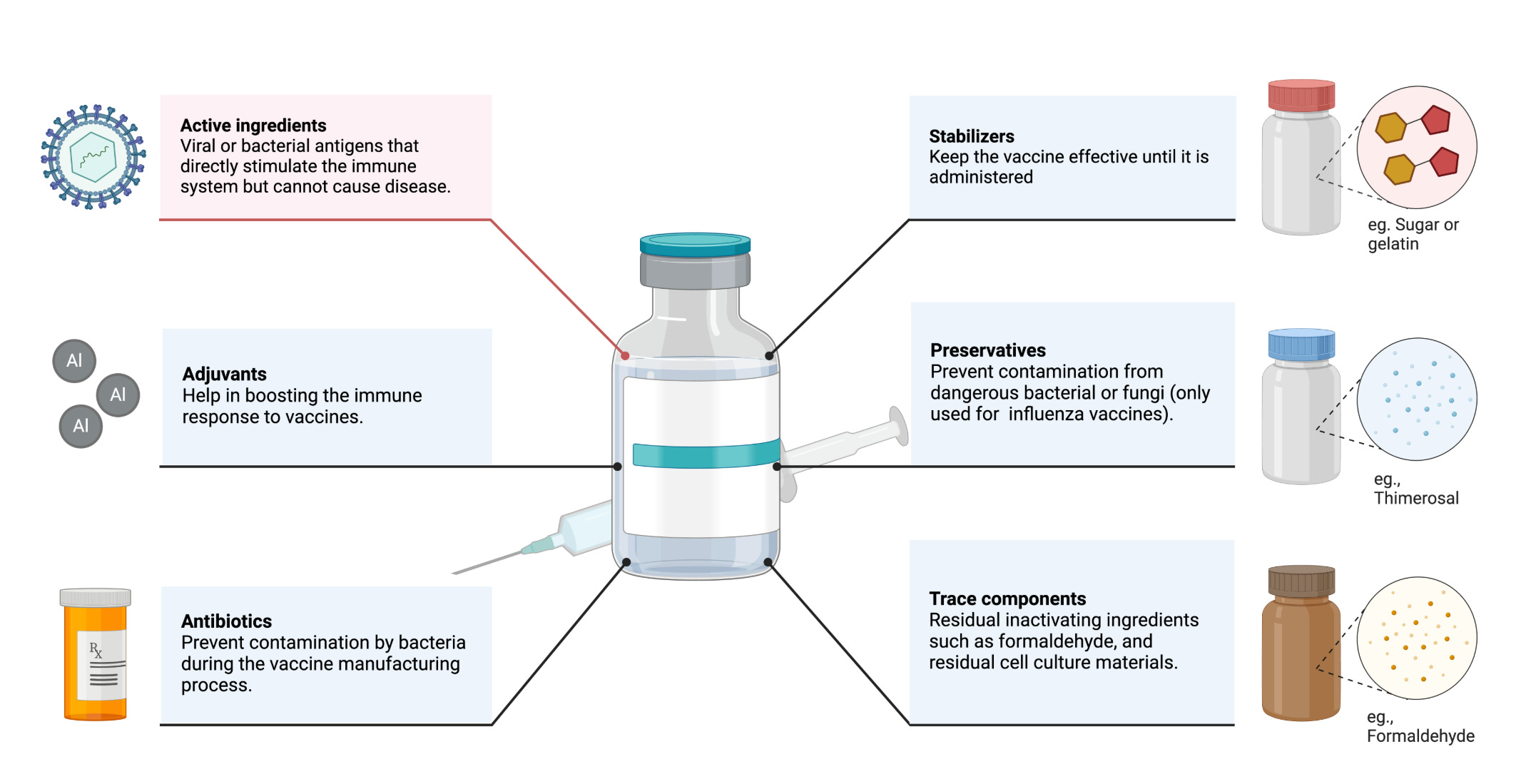
The most important component of vaccines are one or more protein antigens, either directly derived from the pathogen of interest or biomanufactured (15). Polysaccharide antigens also exist and can also induce protective immune responses. They have been the basis of vaccines like pneumonia and meningitis caused by Streptococcus pneumoniae (29). These antigens are responsible for inducing immune responses that provide protection. Other vaccine components may include preservatives (eg. thimerosal, phenol, formaldehyde) to prevent contamination by bacteria or fungi; stabilizers (eg. gelatin, lactose, sorbitol) to keep the vaccine potent during transportation and storage, especially under temperature variations; and excipients (eg. aluminum salts) to ensure vaccine consistency and efficacy (15). Adjuvants are often added to increase potency, reduce the dose of vaccine which must be administered, and increase efficacy in some populations (e.g., infants, elderly, and immunocompromised individuals)(30)(Figure 2).
Adjuvants primarily function by promoting the generation of APCs and co-stimulatory signals through the activation of APCs. After antigen peptides are taken up and processed, APCS present them on their surface via MHCs. Co-stimulatory signals include co-stimulatory molecules (e.g., CD40, CD80, CD86) expressed on the surface of APCs and secreted inflammatory cytokines (e.g., IL-6, IL-10, IL-12, and TNF-α) (31). The production of these two signals can strongly enhance the activation of naïve T cells, thereby boosting the adaptive immune response (32).
There are a few adjuvants that are commonly used in licensed vaccines: aluminum adjuvants (33), MF59 (34), AS03 (35), AS04, CpG ODN 1018, and AS01 (36) are classical human vaccine adjuvants (37)(31). Collectively, these adjuvants act to stimulate pathways that would be stimulated by danger-signals coming from a virus during natural infection. For mRNA vaccines however, the mRNA molecule serves as both an immunogen and adjuvant, due to the intrinsic immunostimulatory properties of mRNA.
Immune responses to mRNA vaccines greatly rely on the delivery system, the immunogenicity of the encoded antigen, and the longevity and subcellular localization of antigen expression. Intramuscular and intradermal administration of mRNA vaccines is highly immunogenic and induces local cytokine and chemokine production that initiates prompt recruitment of neutrophils, monocytes, and other cells to prime the immune response. Injection of mRNa encapsulated in lipid nanoparticles (LNPs) has been shown to induce robust infiltration of neutrophils, monocytes, and dendritic cells as well as the activation of pro-inflammatory cytokines and chemokines in mice (38)(39). LNP formulations are made up four components: ionizable (for example, 1,2-dilinoleyl oxy-N,N-dimethyl-3-aminopropane (40)) or cationic (for example, 1,2-di-O-octadecenyl-3-trimethylammonium-propane (41) and Dimethyldioctadecylammonium bromide (42)) lipids, phospholipids (for example, phosphatidylcholine and phosphatidylethanolamine), cholesterol, and polyethylene glycol (PEG) lipids. These lipids can improve nanoparticle properties, such as particle stability, delivery efficacy, tolerability and biodistribution (43)(12). The most common design of LNPs involves PEG lipids to avoid immune recognition, extend their blood circulation, and improve LNP during synthesis and storage (44-45).
mRNA vaccines have been shown to promote Th1-type immune responses, and potent inductions of antigen-specific germinal center and T-follicular helper cells responses (38)(46-47). Previous research indicates that adjuvant activity of LNPs relies on the ionizable lipid component and IL-6 cytokine induction, rather than MyD88- or MAVS-dependent sensing of LNPs (38). Compared to inactivated or recombinant vaccines, mRNA vaccines using LNP adjuvants induce a stronger and more sustained immune response. This is likely due to the type and amount of cytokines released by the LNP adjuvants as well as prolonged antigen presence in the body up to ten days after intramuscular and intradermal injections (48), allowing for an extended period of immune activation.
mRNA vaccines
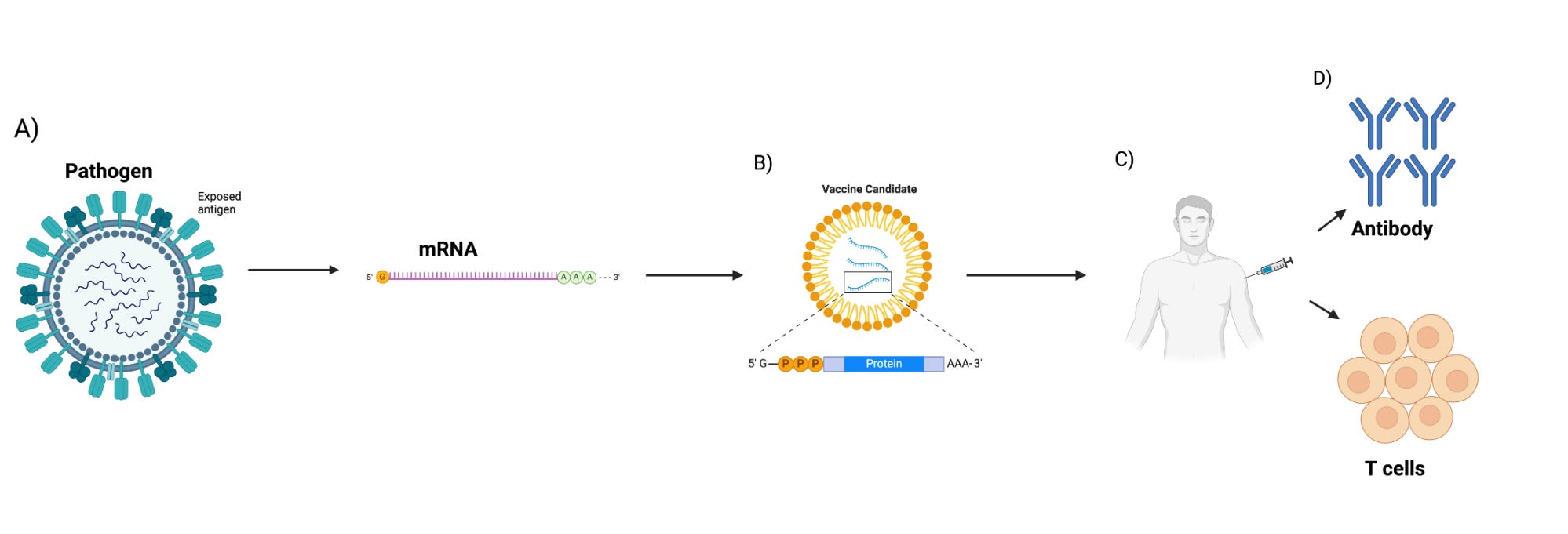
mRNA is a single-stranded ribonucleic acid that is transcribed using DNA as a template and can carry genetic information to guide protein synthesis. The idea of mRNA-based therapeutics emerged over three decades ago when Dimitriadis (49), Malone et al. (41), and Wolff et al. (11) provided the first evidence that mRNA produced in vivo and IVT could be introduced into cells and animals to produce proteins. Major limitations such as potent inflammation and reduced in vivo translation due to mRNA short half-life were quickly recognized. Inflammation-mediated inhibition of protein production, instability of the mRNA molecules, easy degradation by enzymes, and difficulties in delivering mRNA into cells effectively further limited the potential clinical and therapeutic application of the platform (50). Overcoming these shortcomings significantly improved the platform, enabling the successful development of vaccines and adjuvants. Martinon et al. and Conry et al. (51) showed that mRNA loaded into liposomes elicited antigen-specific cytotoxic T lymphocytes and humoral responses, paving the way for mRNA vaccine development and early human trials (Figure 3).
Over the past decade, major technological innovation and research investment have enabled mRNA to become a promising therapeutic tool. Recent advances that were critical for the development of a safe and potent mRNA vaccine platform include the incorporation of modified nucleosides into mRNA (52-53) and the removal of contaminants using purification chromatography (54-55), which were both pioneered by Kariko and Weissman. Improvements in sequence engineering and codon optimization (56), innovations in cap moieties and capping strategies (57), and the evolution of potent and relatively safe delivery systems such as lipid nanoparticles (LNPs) (58) have also significantly advanced the development and regulatory approval of mRNA-based vaccines. Nucleoside modification and elimination of double-stranded RNA contaminants generated during IVT have improved the intrinsic adjuvant effect of the IVT mRNA, improved tolerability, and boosted antigen expression by several folds (59)(52). New methods for adding caps to mRNA molecules have improved how much usable mRNA is produced and reduced its detection by the body's immune system sensors (eg., RIG-I and MDA5), again resulting in improved translation, enhanced safety, and reduced costs. Two mRNA vaccines are currently FDA-approved and are both for the SARS-Cov-2 pathogen (COMIRNATY® and SpikeVax®)(Figure 4).
Pharmacology of mRNA vaccines
mRNA structure is composed of 5’cap, untranslated regions (UTRs), and the poly(A) tail, and the open reading frame (ORF) encoding target antigen (60)(Figure 5).
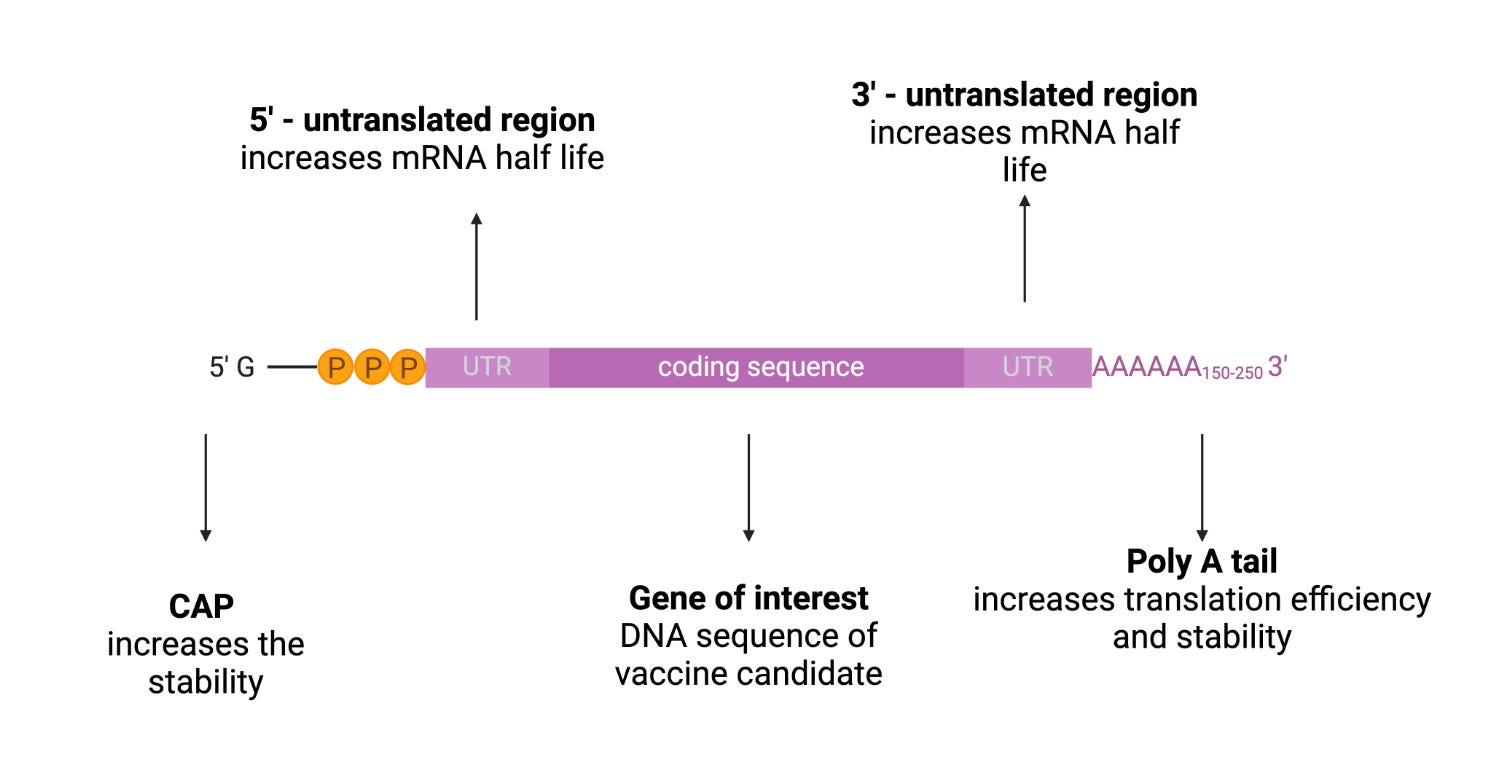
The 5′ end of the mRNA features a 7-methylguanosine (m7G) cap, connected to the first nucleotide by a triphosphate bridge (m7GpppN). m7GpppN is called a 5’ cap, which is a protective structure which protects RNA from exonuclease cleavage, regulates pre-mRNA splicing, and initiates mRNA translation and nuclear export of the mRNA to the cytoplasm (17)(61). The 5’ cap structure can enhance the translation of mRNA. Combining a 5’ cap with eukaryotic translation initiation factor 4e is the key to effective translation (62). The cap is also one of the determinants of mRNA degradation as it regulates the degradation of mRNA, in combination with scavenger decapping enzymes Dcp1, Dcp2, and DcpS (63).
Although UTRs do not encode the desired antigen or a protein, they play a critical role in regulating mRNA expression and stability. These regions are situated between the ORF and the 5′ and 3′ ends (17). They also facilitate mRNA recognition by ribosomes and contribute to post-transcriptional modification of mRNA (64). Inclusion of naturally occurring sequences, such as those derived from alpha- and beta-globins, have been widely used to design mRNA constructs for vaccines (65-66). For example, Zeng et al. developed de novo 5′ UTR sequences based on the guanine–cytosine content and length for mRNA vaccine development. (17)(67).
The IVT mRNA has a polyadenylated section at its 3′ end which is known as the poly(A) tail. The poly(A) tail regulates the stability, translation effectiveness, and lifespan of the mRNA (68). Since the tail size affects the degradation of mRNA, the incorporation of poly(A) tails is desirable in the production of mRNA vaccines and therapeutics with longer half-life. For example, the addition of approximately 100 nucleotides to the poly(A) tail can result in the production of mRNA with desired prolongation of degradation (69)(17).
Natural mRNA contains ATP, CTP, GTP, and UTP as the four basic nucleotides. Although non-modified mRNA has its own advantages, modified nucleotides are beneficial because they can avoid the recognition of IVT mRNA by the innate immune system, avoiding any undesirable immune responses, and improving the translation efficiency of the mRNA (70). Nucleotides that are modified after post-transcriptional modification of mRNA molecules, such as pseudouridine and 5-methylcytidine, can be utilized in the IVT transcription of the mRNA (71). Andries et al. demonstrated that mRNAs containing the N(1)-methyl-pseudouridine (m1Ψ) modification outperformed the pseudouridine (Ψ)-modified mRNA platform by providing up to approximately 44-fold higher and 13-fold higher reporter gene expression upon transfection into cell lines or mice, respectively (72).
Advantages of mRNA vaccines
The use of mRNA has several beneficial features over other vaccines platforms such as subunit, inactivated and live attenuated virus vaccines. Firstly, mRNA is a non-infectious, non-integrating platform, meaning that there is no potential risk of infection or insertional mutagenesis. Additionally, mRNA is degraded by normal cellular processes, and its in vivo half-life can be regulated through various modification and delivery methods (73)(52)(74). The inherent immunogenicity of the mRNA can be down-modulated to further increase the safety profile (73)(75). Secondly, various modifications can make mRNA more stable and highly translatable. For example, efficient in vivo delivery can be achieved by enclosing mRNA within carrier molecules, allowing rapid uptake and expression in the cytoplasm (74)(76). Anti-vector immunity is also avoided because mRNA is the minimal genetic vector, meaning that mRNA vaccines can be administered repeatedly. Third, mRNA vaccines have the potential for rapid, inexpensive and scalable manufacturing, mainly owing to the high yield of in vivo transcription reactions and the fact that mRNA vaccines are produced in a cell-free environment (77). For example, a 5 L bioreactor can produce a million doses of mRNA vaccine in a single reaction (17). Finally, mRNA vaccines have the provision to code for multiple antigens, thus strengthening the immune response against some resilient pathogens (78).
Classes of mRNA vaccines
mRNA vaccines can be divided into three major categories: conventional mRNA, self-amplifying mRNA (SAM), and circular RNA.
Conventional IVT mRNAs are relatively simple in their architecture and are manufactured at high yield using a cell-free template-directed enzymatic synthesis (14). SAM is designed to include viral-derived molecular machines, such as alphavirus enzymes and conserved sequence elements, allowing it to replicate itself within cells after delivery (79). Typical SAM architecture is constructed from an expression cassette that includes a subgenomic promoter and the target antigen. This cassette is then placed between sequences that encode for alphavirus-derived nonstructural proteins and a poly adenosine tail. SAM vaccines are designed with special sequences that include proteins (eg., nsP1-4) which form a RNA-dependent RNA polymerase complex. This complex recognizes specific design elements in the vaccine and replicates the mRNA within the cell cytoplasm. This replication leads to efficient and prolonged transcription and protein expression. However, SAMs are quite large (6,000-12,000 nucleotides) and their manufacturing is more complex and challenging compared with conventional mRNA vaccines due to low yield, difficulty in purification, and are more prone to autocatalysis and physical degradation (80).
Most currently used SAM vaccines are based on an alphavirus genome, where the genes encoding the RNA replication machinery are intact but the genes encoding the structural proteins are replaced with the antigen of interest (77). SAM format cannot easily incorporate modified nucleosides due to interference between the RNA-dependent RNA polymerase and the nucleoside modified sequences, leading to less mRNA being made in target cells (81). The detection of unmodified nucleosides in SAMs by endosomal and cytoplasmic sensors triggers a strong type I interferon response, which poses a challenge for clinical use. However, vaccine dosage of SAMs is significantly lower – about 100 times less – than conventional mRNA vaccines and therefore could offer disease protection with fewer side effects. Another advantage of SAM vaccines is that they create their own adjuvants in the form of dsRNA structures, replication intermediates and other motifs, which may boost their potency. However, the intrinsic nature of these PAMPs may make it difficult to modulate the inflammatory profile or reactogenicity of SAM vaccines. Additionally, SAM vaccines have stricter size constraints than mRNAs that do not encode replicon genes, and the immunogenicity of the replication proteins may theoretically limit repeated use (77). Early experiments developed by the Imperial College and Acuitas Therapeutics used a SAM administered at extremely low doses (10 ng, prime boost), in mice. The experiment showed potent cell and antibody responses in mice (82) and is now being tested at doses 300-1,000x lower than those used in approved nucleoside modified mRNA vaccines (58).
Circular RNA is a class of non-coding single-stranded RNAs found in eukaryotic cells. Circular RNAs are created through a special process called back-splicing, where the ends of the RNA strand join together instead of lining up end-to-end. Due to their stability, long life, low immunogenicity, and translatability, engineering circular RNA has become a vaccine of high interest. Circular RNAs have been engineered to enable protein expression through the addition of internal ribosomal entry sites and/or the incorporation of specific nucleoside modifications in the 5’ UTR (83). This novel platform has proven effective at producing stable and potent translation in eukaryotic cells (84) due to the RNA being more durable and less susceptible to breakdown. Recent studies have suggested that circular RNAs can evade intracellular immune sensors such as RIG-I without nucleoside modifications (83). Qu et al. showed that circular RNA generates potent, antigen-specific CD4+ and CD8+ cellular and humoral immune responses in mice against SARS-CoV-2 and its emerging variants, providing proof of concept for vaccine applications (85). While circular RNA have great potential to become the next generation of RNA-based vaccine platforms, they are at an early stage in terms of design, synthesis, purification, delivery, and application. In general, five pivotal challenges require attention: designing circular RNAs with low immunogenicity and high antigenic yields, improving the circularization efficiency of linear RNA precursors, adequately purifying to avoid contaminants, establishing suitable delivery systems and enabling disease therapeutic applications. For information on methods to specifically improve circular RNA, the reader is encouraged to read this excellent review paper (86).
Limitations and risks of mRNA vaccines
Despite all of their advantages, mRNA vaccines generally face shortcomings with their duration of antibody response, safety, dependence on ultra-low cold chain transport, and high reactogenicity. mRNA vaccines have been shown to elicit potent germinal center immunogenic reactions and T helper cell induction in preclinical studies against HIV-1, SARS-CoV-2 (87-88), Zika virus, and influenza virus (89)(17). Despite these promising results, the duration of the antibody response varies highly from antigen to antigen.
mRNA is inherently unstable in vivo and antigen expression levels are significantly more transient than observed with DNA vaccination, typically only persisting for several days (90). This is at least in part due to viral RNA sensing molecules such as TLRs 3, 7, and 8 as well as Retinoic Acid-Inducible Gene I (RIG-I). Activation of these innate immune sensors triggers a signaling cascade that enhances RNA clearance and also leads to transfected cell apoptosis, limiting antigen production (91-92). Although these events are proinflammatory, it may reduce a potential adaptive immune response due to the suppression of antigen expression.
Consequently, the mRNA delivered in clinically approved SARS-CoV-2 vaccines has been designed to extend the pharmacological half-life of the spike protein. Some reports suggest that introducing highly modified genetic material to patients elicits an immune response that does not adequately resemble natural infection. In fact, some studies demonstrate that the differences in the elicited immune response may be deleterious. Engineering of mRNA vaccine enriches guanine and cytosine content, increased guanine and cytosine content can result in accumulation of G-quadruplex, which could have effects on post-transcriptional regulation via microRNAs, and that defect in post-transcriptional regulation could favor a greater expression of oncogenes, resulting in cancer and neurological diseases.
These distinct differences in cellular expansion and phenotype can be linked to a downregulation of INF I response, which in turn may make patients more susceptible to other infectious diseases or compromise their anti-cancer surveillance. For example, in a study examining the efficacy of OVA-LNP vaccination to induce anti-cancer response against B16-OVA, so and so demonstrated that likelihood of long term survivors was directly related to the degree of modification of the mRNA uridine. They further demonstrated that increasing uridine modification was demonstrably worse for eliciting INF I responses, clearly indicating that mRNA engineering strategies remain nascent and warrant further investigation in the context of globally accessible vaccine products.
In this regard, in the case of SARS-CoV-2 BNT162b2 mRNA vaccine, unlike the immune response induced by natural SARS-CoV-2 infection, where a robust interferon response is observed, those vaccinated with BNT162b2 mRNA vaccines developed a robust adaptive immune response which was restricted only to memory cells, i.e., an alternative route of immune response that bypassed the IFN mediated pathways
Overall, mRNA vaccines have promising safety profiles as supported by clinical trials and real population data. However, there have been some safety incidents that indicate the need for further optimization of mRNA and its components. For instance, CureVac’s protamine-based rabies vaccine, CV7201, caused adverse effects in 78% of participants (93). In Phase I trials of Moderna’s influenza H10N8 vaccine, adverse events were observed from the 400 μg, causing them to use a lower dose of up to 100 μg (94). Furthermore, mild anaphylactic reactions have been seen in 4.7 per million COVID-19 vaccinations, with 2.5 per million vaccinations with the Moderna vaccine and 2.2 per million with Pfizer–BioNTech vaccine (95).
Current mRNA vaccines require improvement, especially in reducing reactogenicity, despite their success against COVID-19. While the reactogenicity may be deemed acceptable for a few doses during a pandemic, there is a demand for mRNA vaccine formulations with milder reactogenicity for repeated boosting against COVID-19 and broader applications in other infectious diseases. Systemic reactogenicity is more common after the second dose of vaccines than the first dose in humans (96), suggesting the involvement of adaptive immunity in reactogenicity. Moreover, enhanced reactogenicity coincided with higher levels of systemic IFN-γ secretion after the second dose in humans and mice (97). In a mouse study, CD4+ and CD8+ T cells were the primary producers of IFN-γ in the second dose, and the administration of an IFN-γ receptor neutralizing antibody dampened the activation of macrophages, monocytes, and dendritic cells. These findings suggest a close relationship between reactogenicity and immunogenicity. However, an IFN-γ receptor neutralizing antibody minimally affected the efficiency of humoral and cellular immunity induction in mice. Furthermore, the intensity of systemic reactogenicity showed limited correlation with the efficacy of humoral and cellular immunity induction in humans (97-98). These results indicate the potential to separate reactogenicity and immunogenicity. Elucidating innate immune signaling pathways responsible for reactogenicity and immunogenicity could benefit the development of effective vaccines with reduced reactogenicity. mRNA delivery systems that efficiently accumulate in the lymph nodes with minimal systemic leakage might mitigate systemic adverse effects while also maintaining effectiveness (97)(99-100).
Affordable and easy access to vaccines is the greatest challenge in achieving prevalent protection against infectious diseases. This access is made further difficult due to the cold storage requirements and stability of mRNA vaccines. mRNA is highly susceptible to RNase enzymes, which degrade it easily (101). Therefore, it requires extreme sterile conditions of an RNase-free environment in preparation, storage, and administration. All equipment used for these three stages must be sterile. On a more local level, portable and reusable Arktek freezers were key in supplying vaccines to over 400,000 people during the 2014-2016 Ebola virus outbreak in West Africa. However, vaccinating billions of people such as during the COVID-19 pandemic requires thermostable vaccines. Two SARS-CoV-2 vaccine candidates were reported to be thermostable in preclinical studies at room temperature (102-103).
Ongoing work
The development of potent and biodegradable lipids, as well as new formulations for the effective delivery of mRNA in vivo will likely address many of these shortcomings. Researchers at the Novartis Institute used the ionizable lipid DLinDMA as the main component of LNPs for a SAM vaccine targeting the respiratory syncytial virus fusion glycoprotein. It was prepared through ethanol dilution and the particle size ranged from 79 to 121 nm. This vaccine technique with an encapsulation rate of 85%-98% elicited a broad, potent, and protective immune response in mice (104). Hoerr et al. combined IVT-mRNA encoding galactosidase with a polycationic peptide (protamine) and then encapsulated the complex in liposomes. The resulting lipopolysaccharide protected the IVT-mRNA for longer and also demonstrated protein expression and subsequent immune response (105). A final example showcasing the potential of delivery vehicles was Geall et al. showcased that nonviral delivery of a 9-kb SAM RNA vaccine encapsulated within an LNP significantly increased immunogenicity compared to delivery of unformulated RNA. The formulation triggered a broad, strong, and protective immune response, comparable to viral delivery methods but without the drawbacks of viral vectors (104).
Exogenous mRNA is inherently immunostimulatory, as it is recognized by a variety of cell surface, endosomal and cytosolic innate immune receptors. This feature is advantageous for vaccination because it may provide adjuvant activity to drive DC maturation and thus elicit robust T and B cell immune responses. However, innate immune sensing of mRNA has also been associated with the inhibition of antigen expression and may negatively affect the immune response (77). Studies over the past decade have shown that the immunostimulatory profile of mRNA can be shaped. Karikó et al. showed that mRNA containing pseudouridines have a higher translational capacity than modified mRNA when tested in mammalian cells and lysates or when administered intravenously into mice at 0.015–0.15 mg/kg doses. The delivered mRNA and the encoded protein could be detected in the spleen at 1, 4, and 24 hours after the injection, where both products were at significantly higher levels when pseudouridine-containing mRNA was administered. These findings indicate that nucleoside modification is an effective approach to enhance stability and translational capacity of mRNA while diminishing its immunogenicity in vivo (52).
Currently, altering mRNA vaccines pharmacokinetic properties by prolonging the translation of antigenic mRNA has emerged as a promising tool to enhance antibody response (90). The use of modified nucleotides such as 5-methyl cytosine or pseudouridine can limit recognition by RIG-I and enables transfected cells to express antigen for significantly longer timeframes (92)(52). Additionally, incorporation of RNA capping analogues have been reported to reduce innate sensing and increase the duration of antigen production in vivo, leading to enhanced immune responses to the encoded antigen (106). Recent clinical trials from Moderna using intramuscular delivery of nucleoside modified RNA show that mRNA vaccines are safe and capable of inducing effective humoral responses against H10N8 and H7N9 influenza strains in humans. In this trial, virus-neutralizing antibody responses were sustained for over 6 months after immunization (94). In studies of recombinant protein vaccines, the exposure kinetics of antigens and adjuvants drastically impact vaccination outcomes, with sustained exposure leading to enhanced antibody production (107-108). Therefore, modulating the kinetics in mRNA vaccines could improve their efficacy. To achieve this, several reports have attempted to control the release of mRNA nanoparticles from hydrogels. For example, mRNA complexed with a lipid-based commercial reagent was loaded onto a poly (2-hydroxyethyl methacrylate)-based porous scaffold (109). After subcutaneous injection, this scaffold retained mRNA for more than 3 days and enhanced protein expression efficiency compared to bolus mRNA injection.
To improve the stability of mRNA vaccines and thus accessibility of mRNA vaccines, mRNA modification of both the coding and non-coding regions can be effective to improve the stability, immunogenicity, and translation efficiency of mRNAs. For example, replacement of uridine with pseudouridine in the coding region reduced degradation by RNase in a nucleoside-modified approach (110). Both Moderna and Pfizer modified the mRNA encoding viral spike protein by replacing natural residues with two consecutive prolines at amino acid positions K986 and V987 for their COVID-19 vaccines (111). The modification allowed stabilization of the spike on the virus particle in its ‘pre-fusion’ conformation. Modifications of the non-coding regulatory regions also increase resistance against degradation by RNase and exonuclease. Studies have also indicated that nucleoside modifications have increased stability of mRNA vaccines. For example, substitution with m1Ψ, m6A, and s2U in mRNA molecules suppresses the degradation of RNA by RNase L (112). Unmodified mRNA can activate RNA-dependent protein kinase, which is one of four kinases that phosphorylate eIF2, a translation initiation factor, leading to the repression of translation (113-114)(86). Therefore, modifying mRNA is necessary to improve vaccine stability. Besides nucleoside modification, common approaches, such as 5′-cap modification and elongation of poly(A) tail, have been applied to optimize mRNA vaccines (115-116). Locked nucleic acid (LNA) caps have also been investigated, in which the ribose is locked in an C3′-endo conformation by a bridging methylene group between the 2′ oxygen and 4′ carbon. Although LNAs have primarily been used in oligonucleotides, mRNAs capped by an LNA analog have recently been demonstrated to have increased translational efficiency and stability (117). Finally, a novel continuous freeze-drying technique based on spin-freezing is being explored as a storage technique for mRNA vaccines. This method has several advantages compared to classical batch freeze-drying including a much shorter drying time and improved process and product quality controlling. Meulewaeter et al. studied the stability of optimally lyophilized mRNA LNPs at 4 °C, 22 °C, and 37 °C and found that transfection properties of lyophilized mRNA LNPs were maintained during at least 12 weeks. This study is one of the first that demonstrates that optimally lyophilized mRNA LNPs can be safely stored at higher temperatures for months without losing their transfection properties (118).
Having discussed mRNA vaccines successes and limitations, I turn now to a consideration of how to design next generation mRNA candidates. Ideal vaccines would be safe, have perfect efficacy, achieve long-lasting immunity, and be low cost. Furthermore, they would mimic the endogenous immune response as much as possible in both space and time. This requires sustained locoregional delivery of both the antigen (or antigen encoding sequence) and co-stimulatory molecules to drive the desired immune response. In natural immunity, for example when we are naturally exposed to a virus, the antigen and immunostimulatory molecules (DAMPS, PAMPs, etc) are co-presented to the immune system, demonstrating that mounting both cellular immunity (driven largely by interferon responses and cellular cytotoxicity) and humoral immunity requires a tightly orchestrated immune cascade….
One proposal would be to functionalize the liposomal nanoparticles in which mRNA vaccines are currently delivered with co-stimulatory antibodies such as anti-CD28 and anti-OX40. Alternatively, co-delivery with adjuvants could rationally skew the immune response toward either a Th1 or Th2 type.
Moreover, many groups have been exploring the use of biomaterials to improve vaccine responses. Sustained exposure to mRNA LNPs may, in fact, generate longer lasting germinal centers, in turn allowing for more somatic hypermutation and affinity maturation of B-cells, resulting in better quality antibodies that provide more durable and broad protection against highly mutagenic viruses.
Another approach could utilize a biomaterial scaffold loaded with specific chemokines or cytokines to create an immunological niche in which cells interact with the mRNA loaded liposomal nanoparticles. The hypothesis here is that by changing the context in which cells uptake mRNA and express the encoded protein could alter the downstream immunity generated.
Conclusion
Vaccines continue to be critically important technologies for the protection of public health. They have a long and storied history from early biological and immunological theory in the later 1700s to present day. Decades of research into mRNA design and its delivery have made mRNA vaccines an astonishing tool for combating pandemics and infectious diseases. The recent COVID-19 pandemic has especially catalyzed the development of mRNA based therapeutics. It is evident that mRNA technology has the potential for the development of more effective vaccines against persistent and challenging pathogens. mRNA vaccines offer several advantages, including the ability to induce strong and sustained immune responses, reduced risk of insertional mutagenesis, and the potential for rapid adaptation to emerging pathogens. Nevertheless, advancement in mRNA delivery technologies will be required for more effective, safer, and cold-chain-free mRNA vaccines, having the capacity to vaccinate billions of populations across boundaries. The further incorporation of modified nucleosides, advanced purification methods, and innovative delivery systems like lipid nanoparticles have significantly enhanced the efficacy and safety of mRNA vaccines. These advancements not only address some of the inherent limitations of the vaccine platform but also open up new avenues for rapid, scalable, and cost-effective vaccine production.
Looking forward, continued research and development in vaccine technologies are essential to further improve their efficacy, safety, and accessibility. Addressing challenges that will come with developing mRNA vaccine platforms will be crucial in creating next-generation vaccines that are more biomimetic, durable, and potent, ultimately ensuring better preparedness for future infectious disease outbreaks and contributing to global health security.
Abbreviations
mRNA: messenger RNA, IVT: in vitro transcribed, B cells: B-lymphocytes cells, CD8+ T cells: cytotoxic T-lymphocytes, APCs: antigen-presenting cells, MHC-I: class I major histocompatibility complexes, TCR: T cell receptors, MHC-II: class II major histocompatibility complexes, CD4+ T cells: T helper cells, DCs: dendritic cell, PAMPs: pathogen associated molecular patterns, PRRs: pattern recognition receptors, TLRs: Toll-like receptors, Th1: T helper 1 cell, Th2: T helper 2 cell, IFN-γ: Interferon-gamma, MBCs: memory B cells, Tfh: T follicular helper cells, GC: germinal center, LLPCs: long-lived plasma cells, LNPs: lipid nanoparticles, PEG: polyethylene glycol, UTRs: untranslated regions, ORF: open reading frame, m7G: 7-methylguanosine, SAM: self-amplifying mRNA, RIG-I: Retinoic Acid-Inducible Gene I, LNA: Locked nucleic acid
If you truly made it to the end. I can’t thank you enough for the time. If you have any feedback it would be much appreciated. Until next time!
References
Saleh A, Qamar S, Tekin A, Singh R, Kashyap R. Vaccine Development Throughout History. Cureus, 13: 2021.
Pindyck T, Tate JE, Parashar UD. A decade of experience with rotavirus vaccination in the United States – vaccine uptake, effectiveness, and impact. Expert Rev Vaccines, 17: 593-606, 2018.
Sánchez-Sampedro L, Perdiguero B, Mejías-Pérez E, García-Arriaza J, Pilato MD, Esteban M. The Evolution of Poxvirus Vaccines. Viruses, 7: 1726-1803, 2015.
Delany I, Rappuoli R, Gregorio ED. Vaccines for the 21st century. EMBO Molecular Medicine, 6: 708-720, 2014.
Travieso T, Li J, Mahesh S, Blasi M. The use of viral vectors in vaccine development. Nature Vaccines, 7: 75, 2022.
Mohsen Mo, Bachmann MF. Virus-like particle vaccinology, from bench to bedside. Nature Cellular & Molecular Immunology volume, 19: 993-1011, 2022.
Suryawanshi YR. An overview of protein-based SARS-CoV-2 vaccines. Vaccine, 41: 6174-6193, 2023.
Gupta S, Pellett S. Recent Developments in Vaccine Design: From Live Vaccines to Recombinant Toxin Vaccines. Toxins (Basel), 9: 563, 2023.
Cobb M. Who discovered messenger RNA?. Cell Press, 25: R526-R532, 2015.
Jirikowski GF, Sanna PP, Maciejewski-Lenoir Dominique, Bloom FE. Reversal of Diabetes Insipidus in Brattleboro Rats: Intrahypothalamic Injection of Vasopressin mRNA. Science, 255: 996-998, 1992.
Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A, Felgner PL. Direct gene transfer into mouse muscle in vivo. Science, 247: 1465-1468, 1990.
Hou X, Zaks T, Langer R, Dong Y. Lipid nanoparticles for mRNA delivery. Nature Reviews Materials, 6: 1078-1094, 2021.
Embgenbroich M, Burgdorf S. Current Concepts of Antigen Cross-Presentation. Front Immunol, 9: 2018.
Ghattas M, Dwivedi G, Lavertu M, Alameh MG. Vaccine Technologies and Platforms for Infectious Diseases: Current Progress, Challenges, and Opportunities. Vaccines (Basel), 9: 2021
Pollard AJ, Bijker ME. A guide to vaccinology: from basic principles to new developments. Nature Reviews Immunology, 2: 83-100, 2021.
Lazzaro S, Giovani C, Mangiavacchi S, Magini D, Maione D, Baudner B, Geall AJ, Gregorio ED, D’Oro U, Buonsanti C. CD8 T-cell priming upon mRNA vaccination is restricted to bone-marrow-derived antigen-presenting cells and may involve antigen transfer from myocytes. Immunology, 2: 312-326, 2015.
Gote V, Bolla PK , Kommineni N, Butreddy A,Nukala PK, Palakurthi SS, Khan W. A Comprehensive Review of mRNA Vaccines. Int J Mol Sci, 24: 2700, 2023.
Kim J, Eygeris Y, Gupta M, Sahay G. Self-assembled mRNA vaccines. Adv Drug Deliv Rev, 170: 83-112, 2021.
Rijkers GT, Weterings N, Obregon-Henao A, Lepolder M, Dutt TS, van Overveld FJ, Henao-Tamay M. Antigen Presentation of mRNA-Based and Virus-Vectored SARS-CoV-2 Vaccines. Vaccines (Basel), 8: 848, 2021.
Amarante-Mendes, GP, Adjemian S, Branco LM, Zanetti LC, Weinlich R, Bortoluci KR. Pattern recognition receptors and the host cell death molecular machinery. Frontiers in Immunology, 9: 2379, 2018.
Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduction and Targeted Therapy, 6: 291, 2021.
Mogensen TH. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin Microbiol Rev, 2: 240-273, 2009.
Elena Cano, Luz R; Damaris, H; Lopera, E., Autoimmunity: From Bench to Bedside. El Rosario University Press, Bogota, Colombia, 2013.
Chauveau A, Pirgova G, Cheng H, De Martin A, Zhou FY, Wideman S, Rittscher J, Ludewig B, Arnon TL. Visualization of T Cell Migration in the Spleen Reveals a Network of Perivascular Pathways that Guide Entry into T Zones. Immunity, 5: 794-804, 2020.
Crotty S. T follicular helper cell biology: A decade of discovery and diseases. Immunity, 5: 1132-1148, 2019
Griffiths GM, Berek C, Kaartinen M, Milstein C. Somatic mutation and the maturation of immune response to 2-phenyl oxazolone. Nature, 312: 271-275, 1984.
McKean D, Huppi K, Bell M, Staudt L, Gerhard W, Weigert M. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. PNAS, 10: 3180-3184, 1984.
Akkaya M, Kwak K, Pierce SK. B cell memory: building two walls of protection against pathogens. Nature Reviews Immunology, 20: 229-238, 2020
Robbins JB, Schneerson R, Szu SC, Fattom A, Yang Y, Lagergard T, Chu C, Sørensen US. Prevention of invasive bacterial diseases by immunization with polysaccharide-protein conjugates. Current Topics in Microbiology and Immunology, 146: 169-180, 1989.
Coffman RL, Sher A, Seder RA. Vaccine Adjuvants: Putting Innate Immunity to Work. Immunity, 33: 492-503, 2010.
Z T, C Y, Jiang Y, He X, Wei Y, Yu Y, Tian X. Vaccine adjuvants: mechanisms and platforms. Signal Transduction and Targeted Therapy, 8: 283, 2023.
Janeway C. Immunogenicity signals 1,2,3... and 0. Immunology Today, 10: 283-286, 1989
Ravi Danielsson R, Eriksson H. Aluminium adjuvants in vaccines – A way to modulate the immune response. Seminars in Cell & Developmental Biology, 115: 3-9, 2021.
Koa E, Kang S. Immunology and efficacy of MF59-adjuvanted vaccines. Hum Vaccin Immunother, 12: 3041-3045, 2018.
Grigoryan L, Feng Y, Bellusci L, Lai L, Wali B, Ellis M, Yuan M, Arunachalam PS, Hu M, Kowli S, Gupta S, Maysel-Auslender S, Maecker HT, Samaha H, Rouphael N, Wilson IA, Moreno AC, Suthar MS, Khurana S, Pillet S, Charland N, Ward BJ, Pulendran B. AS03 adjuvant enhances the magnitude, persistence, and clonal breadth of memory B cell responses to a plant-based COVID-19 vaccine in humans. Sci Immunol, 9: 2024.
Coccia M, Collignon C, Hervé C, Chalon A, Welsby I, Detienne S, et al. Cellular and molecular synergy in AS01-adjuvanted vaccines results in an early IFNγ response promoting vaccine immunogenicity. npj Vaccines, 2: 25, 2017.
Xie C, Yao R, Xia X. The advances of adjuvants in mRNA vaccines. npj Vaccines, 8: 162, 2023.
Alameh M, Tombácz I, Bettini E, Lederer K, Ndeupen S, Sittplangkoon C, Wilmore JR, Gaudette BT, et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity, 12: 2877–2892, 2021.
Ndeupen S, Qin Z, Jacobsen S, Bouteau A, Estanbouli H, Igyártó BZ. The mRNA-LNP platform's lipid nanoparticle component used in preclinical vaccine studies is highly inflammatory. iScience, 12: 103479, 2021.
Heyes J, Palmer L, Bremner K, MacLachlan I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. Journal of Controlled Release, 107: 276-287, 2005.
Malone RW, Felgner PL, Verma IM. Cationic liposome-mediated RNA transfection. PNAS, 16: 6077-6081, 1989.
Loug G, Anderluzzi G, Schmidt ST, Woods S, Gallorini S, Brazzoli M, et al. Delivery of self-amplifying mRNA vaccines by cationic lipid nanoparticles: The impact of cationic lipid selection. Journal of Controlled Release, 325: 370-379, 2020
Hajj KA, Whitehead KA. Tools for translation: non-viral materials for therapeutic mRNA delivery. Nature Reviews Materials, 2: 17056, 2017.
Kulkarni JA, Witzigmann D, Leung J, Tam YYC, Cullis PR. On the role of helper lipids in lipid nanoparticle formulations of siRNA. Nanoscale, 11: 21733-21739, 2019
Waggoner LE, Miyasaki KF, Kwon EJ. Analysis of PEG-lipid anchor length on lipid nanoparticle pharmacokinetics and activity in a mouse model of traumatic brain injury. Biomater. Sci, 11: 4238-4253, 2023.
Lindgren G, Ols S, Liang F, Thompson EA, Lin A, Hellgren F, Bahl K, John S, Yuzhakov O, Hassett KJ, Brito LA, et al. Induction of Robust B Cell Responses after Influenza mRNA Vaccination Is Accompanied by Circulating Hemagglutinin-Specific ICOS+ PD-1+ CXCR3+ T Follicular Helper Cells. Front Immunol, 8: 1539, 2017.
Pardi N, Secreto AJ, Shan X, Debonera F, Glover J, Yi Y, Muramatsu H, Ni H, Mui BL, Tam YK, et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nature Communications, 18: 14630, 2017.
Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, Madden TD, Hope MJ, Weissman D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J Control Release, 217: 345-351, 2015.
Dimitriadis GJ. Translation of rabbit globin mRNA introduced by liposomes into mouse lymphocytes. Nature, 274: 923–924, 1978.
Alameh, MG. Weissman, D. Pardi, N., Messenger RNA-Based Vaccines Against Infectious Diseases, in Yu D; Petsch B (eds), mRNA Vaccines. Current Topics in Microbiology and Immunology. vol 440., Springer International Publishing, Switzerland, AG, pp 111–145, 2020.
Conry RM, LoBuglio AF, Wright M, Sumerel L, Pike MJ, Johanning F, Benjamin R, Lu D, Curiel DT. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res, 7: 1397- 400, 1995.
Karikó K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S, Weissman D. Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability. Molecular Therapy, 16: 1833-1840, 2008.
Karikó K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity, 2; 165-175, 2005.
Karikó K, Muramatsu H, Ludwig J, Weissman D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Research, 39: 142, 2011.
Baiersdörfer M, Boros G, Muramatsu H, Mahiny A, Vlatkovic I, Sahin U,Karikó K. A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA. Molecular Therapy - Nucleic Acids, 15: 26-35, 2019.
Asrani KH, Farelli JD, Stahley MR, Miller RL, Cheng CJ, Subramanian RR, Browna JM. Optimization of mRNA untranslated regions for improved expression of therapeutic mRNA. RNA Biology, 6: 756–762, 2018.
Trotman JB, Schoenberg DR. A Recap of RNA Recapping. Wiley Interdiscip Rev RNA, 10: 1504, 2018.
Chaudhary N, Weissman D, Whitehead KA. mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nature Reviews Drug Discovery, 20: 817-838, 2021.
Weissman, D. Pardi, N. Muramatsu, H. Karikó, K., HPLC Purification of In Vitro Transcribed Long RNA, In: Rabinovich, P. (eds) Synthetic Messenger RNA and Cell Metabolism Modulation. Human Press, Methods in Molecular Biology, Totowa, NJ, pp 43-54, 2012.
Sahin U, Karikó K, Türeci Ö. mRNA-based therapeutics — developing a new class of drugs. Nature Review Drug Discovery, 13: 759-780, 2014.
Ramanathan A, Robb GB, Chan S. mRNA capping: biological functions and applications. Nucleic Acids Research, 16: 7511- 7526, 2016.
Yang L, Tang L,Zhang M, Liu C. Recent Advances in the Molecular Design and Delivery Technology of mRNA for Vaccination Against Infectious Diseases. Frontiers Immunology, 13: 2022.
Li Y, Kiledjian M. Regulation of mRNA decapping. Wiley Interdisciplinary Review RNA, 1: 253-265, 2010.
Chatterjee S, Pal JK. Role of 5'- and 3'-untranslated regions of mRNAs in human diseases. Biology of the Cell, 101: 251-262, 2008.
Carralot JP, Probst J, Hoerr I, Scheel B, Teufel R, Jung G, Rammensee HG, Pascolo S. Polarization of immunity induced by direct injection of naked sequence-stabilized mRNA vaccines. Cellular and Molecular Life Sciences, 61: 2418–2424, 2004.
Babendure JR,1, Babendure JL,Ding J, Tsien RY. Control of mammalian translation by mRNA structure near caps. RNA, 12: 851-861, 2006.
Zeng C, Hou X, Yan J, Zhang C, Li W, Zhao W, Du S, Dong Y. Leveraging mRNA Sequences and Nanoparticles to Deliver SARS-CoV-2 Antigens In Vivo. Advanced Materials, 32: 2020
Gallie DR. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes and Development, 5: 2108-2116, 1991.
Godiska R, Mead D, Dhodda V, Wu C, Hochstein R, Karsi A, Usdin K, Entezam A, Ravin N. Linear plasmid vector for cloning of repetitive or unstable sequences in Escherichia coli. Nucleic Acids Research, 38: 88, 2010.
Anderson BR, Muramatsu H, Nallagatla SR, Bevilacqua PC, Sansing LH, Weissman D, Karikó K. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Research, 38: 5884-5892, 2010.
Karikó K, Weissman D. Naturally occurring nucleoside modifications suppress the immunostimulatory activity of RNA: implication for therapeutic RNA development. Current Opinion in Drug Discovery and Development, 10: 523-532, 2007.
Andries O, Cafferty SMc, De Smedt SC, Weiss R, Sanders NN, Kitada T. N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. Journal of Controlled Release, 217: 337-344, 2015.
Thess A, Grund S, Mui BL, Hope MJ, Baumhof P, Fotin-Mleczek M, Schlake T. Sequence-engineered mRNA Without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Molecular Therapy, 23: 1456-1464, 2015.
Guan S, Rosenecker J. Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems. Nature Gene Therapy, 24: 133-143, 2017.
Karikó K, Muramatsu H, Ludwig J, Weissman D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Research, 39: 142, 2011.
Kauffman KJ, Webber MJ, Anderson DG. Materials for non-viral intracellular delivery of messenger RNA therapeutics. Journal of Controlled Release, 240: 227-234, 2016.
Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines — a new era in vaccinology. Nature Reviews Drug Discovery, 17: 261-279, 2018.
Freyn AW,da Silva JR, Rosado VC, Bliss CM, Pine M, Mui BL, Tam YK, Madden TD, Ferreira LCS, Weissman D, et al. A Multi-Targeting, Nucleoside-Modified mRNA Influenza Virus Vaccine Provides Broad Protection in Mice. Molecular Therapy, 28: 1569-1584, 2020.
Magini D, Giovani C, Mangiavacchi S, Maccari S, Cecchi R, Ulmer JB, De Gregorio E, Geall AJ, Brazzoli M, Bertholet S. Self-Amplifying mRNA Vaccines Expressing Multiple Conserved Influenza Antigens Confer Protection against Homologous and Heterosubtypic Viral Challenge. PLOS One, 11: 2016.
Bloom K, van den Berg F, Arbuthnot P. Self-amplifying RNA vaccines for infectious diseases. Nature Gene Therapy, 28: 117-120, 2021.
Maruggi G, Zhang C, Li J, Ulmer JB, Yu D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Molecular Therapy, 10. 757-772, 2019.
McKay PF, Hu K, Blakney AK, Samnuan K, Brown JC, Penn R, Zhou J, Bouton CR, Rogers P, Polra K, Lin PJC, Barbosa C, Tam YK, Barclay WS, Shattock RJ. Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nature Communications, 11: 2020.
Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nature Reviews Genetics, 20: 675-691, 2019.
Zhou W, Cai Z, Liu J, Wang D, Ju H, Xu R. Circular RNA: metabolism, functions and interactions with proteins. Molecular Cancer, 19: 172, 2020.
Qu S, Yang X, Li X, Wang J, Gao Y, Shang R, Sun W, Dou K, Li H. Circular RNA: A new star of noncoding RNAs. Cancer Letters, 365: 141-148, 2015.
Niu D, Wu Y, Lian J. Circular RNA vaccine in disease prevention and treatment. Nature Signal Transduction and Targeted Therapy, 8: 2023.
Corbett KS, Flynn B, Foulds KE, Francica JR, Boyoglu-Barnum S, Werner AP, Flach B, O’Connell S, Bock KW, et al. Evaluation of the mRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. The New England Journal of Medicine, 383: 1544-1555, 2020.
Lederer K,1, Castaño D, Atria DG, Oguin TH, Wang S, Manzoni TB , Muramatsu H, Hogan MJ, Amanat F, Cherubin P, et al. SARS-CoV-2 mRNA Vaccines Foster Potent Antigen-Specific Germinal Center Responses Associated with Neutralizing Antibody Generation. Immunity, 53: 1281-1295, 2020.
Pardi N, Hogan MJ, Naradikian MS, Parkhouse K, Cain DW, Jones L, Moody MA, Verkerke HP, Myles A, Willis E, et al. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. Journal of Experimental Medicine, 215: 1571–1588, 2018.
Irvine DJ, Aung A, Silva M. Controlling timing and location in vaccines. Advanced Drug Delivery Reviews, 158: 91–115, 2020.
Besch R, Poeck H, Hohenauer T, Senft D, Häcker G, Berking C, Hornung V, Endres S, Ruzicka T, Rothenfusser S, Hartmann G. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon–independent apoptosis in human melanoma cells. The Journal of Clinical Investigation, 119: 2399–2411, 2009.
Anderson BR, Muramatsu H, Jha BK,3 Silverman RH, Weissman D, Karikó K. Nucleoside modifications in RNA limit activation of 2′-5′-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Research, 39: 9329- 9338, 2011.
Alberer M, Gnad-Vogt U, Hong HS, Mehr KT, Backert L, Finak G, Gottardo R, Bica MA, Garofano A, Koch SD, Fotin-Mleczek M, Hoerr I, Clemens R, Sonnenburg F. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. The Lancet, 390: 1511-1520, 2017.
Feldman RA, Fuhr R, Smolenov I, Ribeiro AM, Panther L, Watson M, Senn JJ, Smith M, Almarsson A, et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine, 37: 3326-3334, 2019.
Shimabukuro TT, Cole M, Su JR. Reports of Anaphylaxis After Receipt of mRNA COVID-19 Vaccines in the US—December 14, 2020-January 18, 2021. JAMA Insights, 325: 1101-1102, 2021.
Baden LR, Sahly HM, Essink B, Kotloff K, Frey S, Novak R, Diemert D, Spector SA, Rouphael N, Creech CB, McGettigan J, Khetan S, et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. The New England Journal of Medicine, 384: 403-416, 2021.
Mochida Y, Uchida S. mRNA vaccine designs for optimal adjuvanticity and delivery. RNA Biology, 21: 1-27, 2024.
Debes AK, Xiao S, Colantuoni E, Egbert ER, Caturegli P, Gadala A, Milstone AM. Association of Vaccine Type and Prior SARS-CoV-2 Infection With Symptoms and Antibody Measurements Following Vaccination Among Health Care Workers. JAMA Internal Medicine, 181: 1660-1662, 2021.
Yang R, Deng Y, Huang B, Huang L, Lin A, Li Y, Wang W, Liu J, Lu S, Zhan Z, Wang Y, Ruhan A, Wen W, Niu P, et al. A core-shell structured COVID-19 mRNA vaccine with favorable biodistribution pattern and promising immunity. Nature Signal Transduction and Targeted Therapy, 6: 2021.
Han X, Alameh M, Butowska K, Knox JJ, Lundgreen K, Ghattas M, Gong N, Xue L, Xu Y, Lavertu M, et al. Adjuvant lipidoid-substituted lipid nanoparticles augment the immunogenicity of SARS-CoV-2 mRNA vaccines. Nature Nanotechnology, 18: 1105–1114, 2023.
Pascolo S. Synthetic Messenger RNA-Based Vaccines: From Scorn to Hype. Viruses, 13: 270, 2021.
Zhang N, Li X, Deng Y, Zhao H, Huang Y, Yang G, Huang W, Gao P, Zhou C, et al. A Thermostable mRNA Vaccine against COVID-19. Cell, 182: 1271–1283, 2020.
Qi Y, Fox CB. Development of thermostable vaccine adjuvants. Expert Review of Vaccines, 20: 497-517, 2021.
Geall AJ, Verma A, Otten GR, Shaw CA, Hekele A, Banerjee K, Cu Y, Beard CW, Brito LA, Krucker T, O'Hagan DT, Singh M, Mason PW, et al. Nonviral delivery of self-amplifying RNA vaccines. PNAS, 109. 14604–14609, 2012.
Hoerr I, Obst R, Rammensee HG, Jung G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. European Journal of Immunology, 30: 1-7, 2000.
Kuhn AN, Diken M, Kreiter S, Selmi A, Kowalska J, Jemielity J, Darzynkiewicz E, Huber C, Türec Oi, Sahin U. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Nature Gene Therapy, 17: 961-971, 2010.
Tam HH, Melo MB, Kang M, Pelet JM, Ruda VM, Foley MH, Hu JK, Kumari S, Crampton J, Baldeon AD, Sanders RW, et al. Sustained antigen availability during germinal center initiation enhances antibody responses to vaccination. PNAS, 113: 6639-6648, 2016.
Cirelli KM, Carnathan DG, Nogal B, Martin JT, Rodriguez OL, Upadhyay AA, Enemuo CA, Gebru EH, Choe Y, Viviano F, Nakao C, et al. Slow Delivery Immunization Enhances HIV Neutralizing Antibody and Germinal Center Responses via Modulation of Immunodominance. Cell, 117: 1153-1171, 2019.
Chen R, Zhang H, Yan J, Bryers JD. Scaffold-mediated delivery for non-viral mRNA vaccines. Nature Gene Therapy, 25: 556-567, 2018.
Rein Verbeke R, Lentacker I, De Smedt SC, Dewitte H. Three decades of messenger RNA vaccine development. Nano today, 28: 2019.
Uddin MN, Roni MA. Challenges of Storage and Stability of mRNA-Based COVID-19 Vaccines. Vaccines (Basel), 9: 1033, 2021.
Anderson BR, Muramatsu H, Jha BK, Silverman RH, Weissman D, Karikó K. Nucleoside modifications in RNA limit activation of 2′-5′-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Research, 39: 9329–9338, 2011.
Sonenberg N, Hinnebusch AG. Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell, 136: 731-745, 2009.
Anderson BR, Muramatsu H, Nallagatla SR, Bevilacqua PC, Sansing LH, Weissman D, Karikó K. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Research, 38: 5884- 5892, 2010.
Kim SC, Sekhon SS, Shin W, Ahn G, Cho B, Ahn J, Kim Y. Modifications of mRNA vaccine structural elements for improving mRNA stability and translation efficiency. Molecular and Cellular Toxicology, 18; 1-8, 2021.
Warminski M, Mamot A, Depaix A, Kowalska J, Jemielity J. Chemical Modifications of mRNA Ends for Therapeutic Applications. Accounts of Chemical Research, 56: 2814-2826, 2023.
Senthilvelan A, Vonderfecht T, Shanmugasundaram M, Pal I, Potter J, Kore AR. Trinucleotide Cap Analogue Bearing a Locked Nucleic Acid Moiety: Synthesis, mRNA Modification, and Translation for Therapeutic Applications. Organic Letters, 23: 4133-4136, 2021.
Meulewaeter S, Nuytten G,MHY, De Smedt SC, Cullis PR, De Beer T, Lentacker I, Verbeke R. Continuous freeze-drying of messenger RNA lipid nanoparticles enables storage at higher temperatures. Journal of Controlled Release, 357: 149-160, 2023.